

Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes
- PMID: 38191930
- PMCID: PMC10864185
- DOI: 10.1038/s41592-023-02124-2
Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes
Abstract
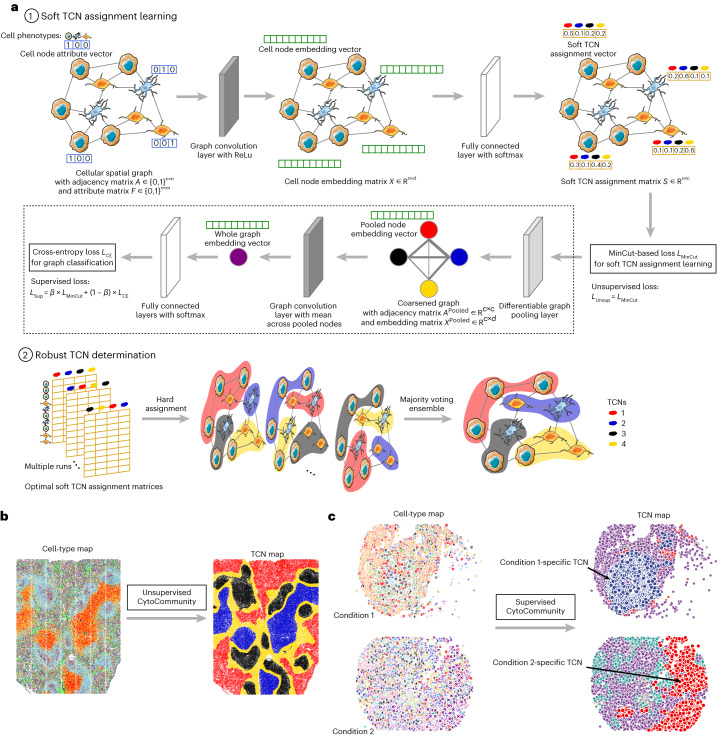
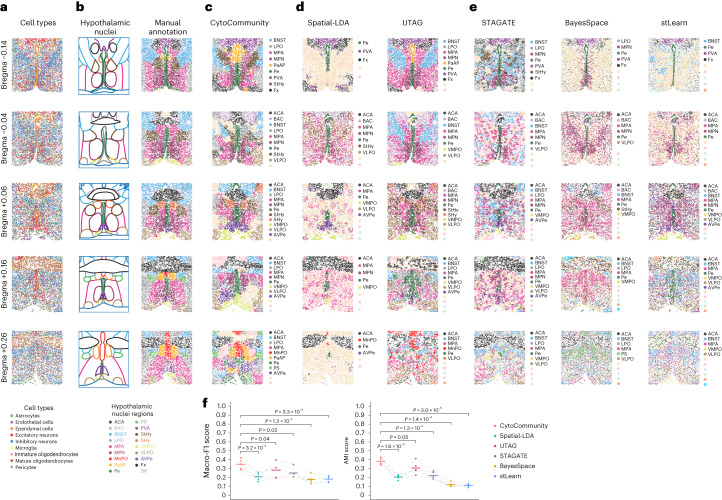
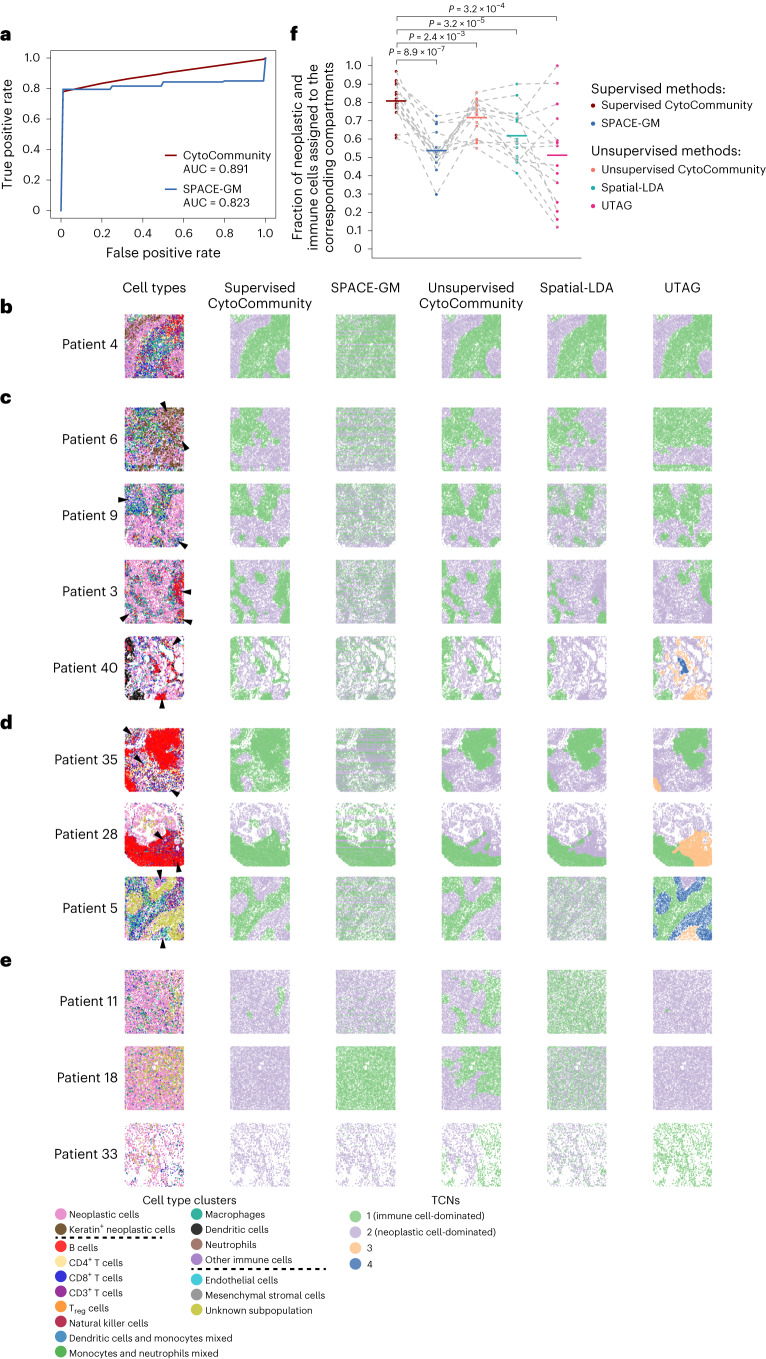
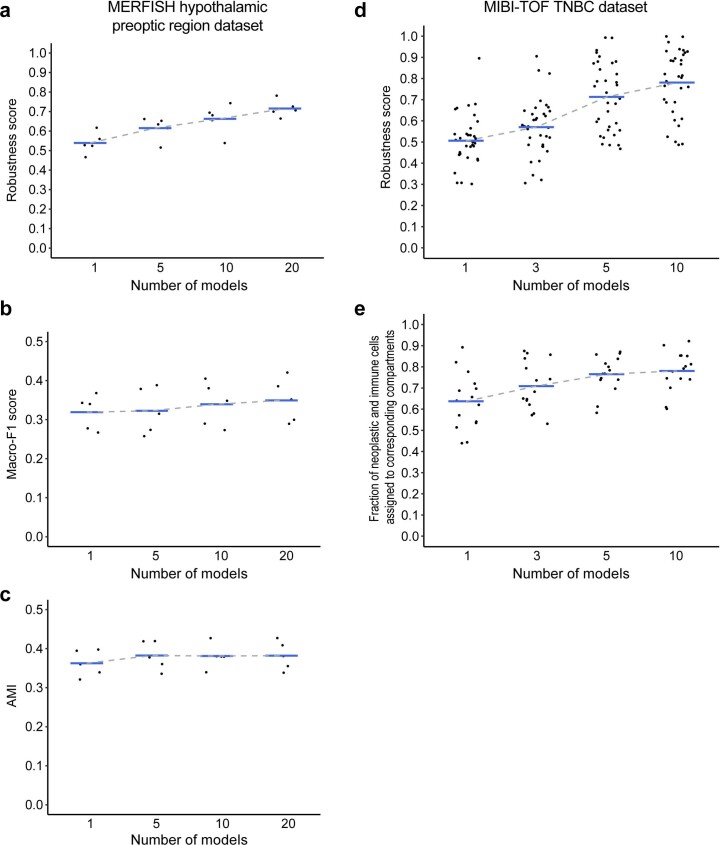
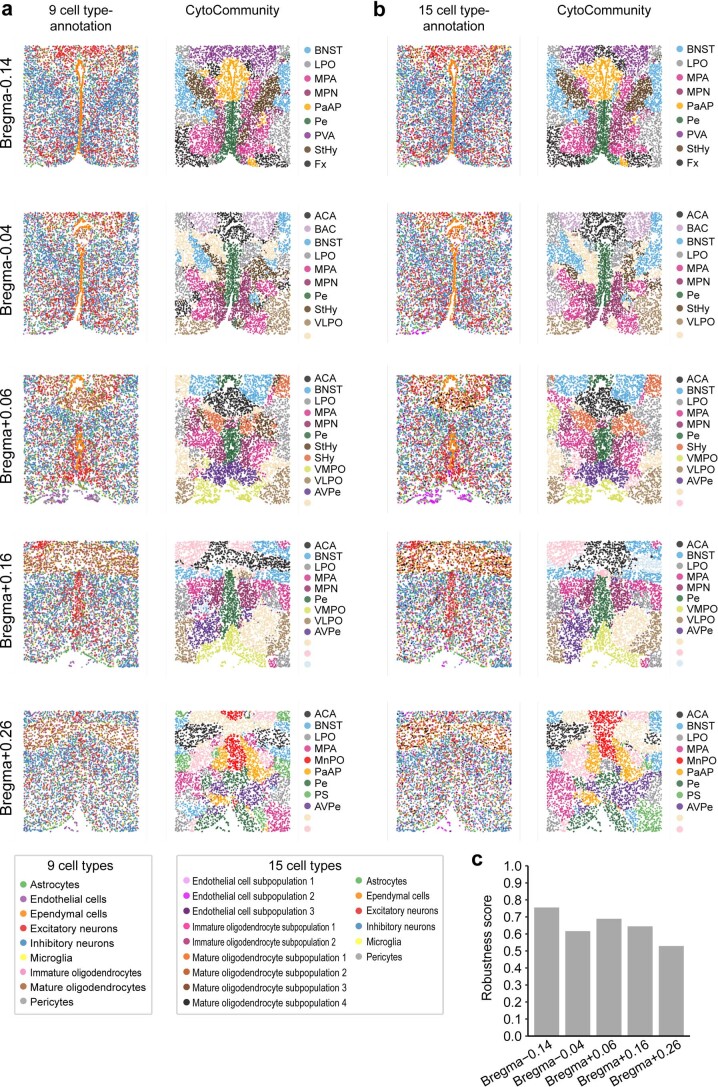
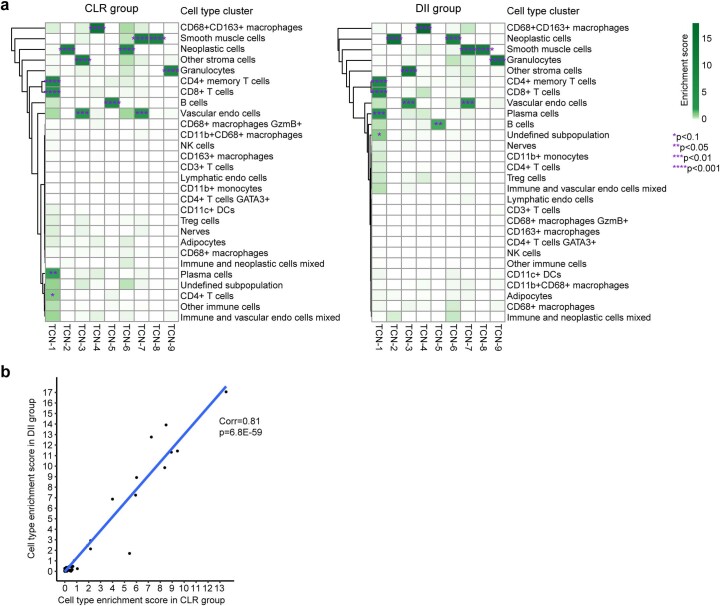
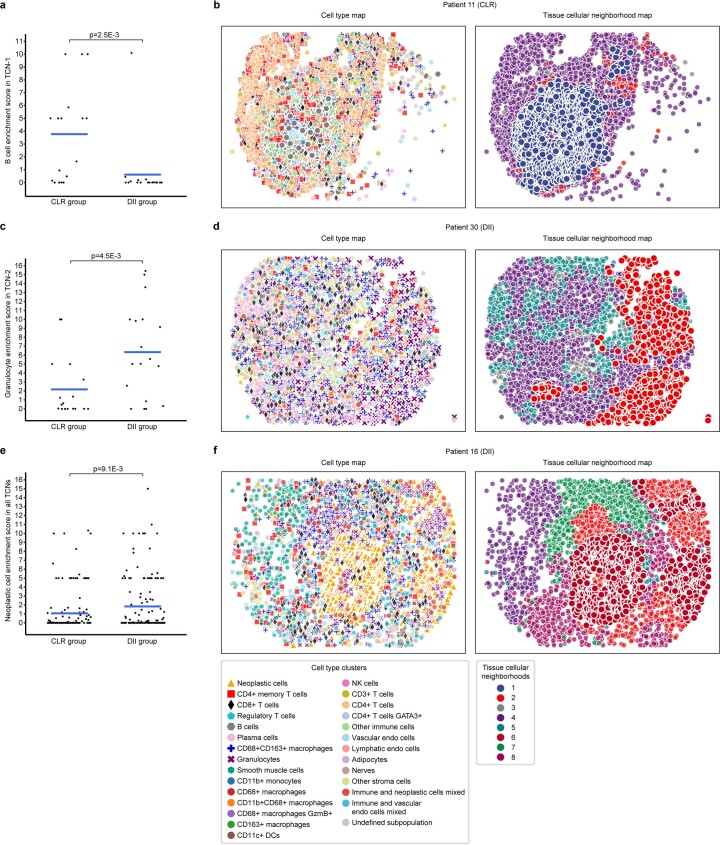
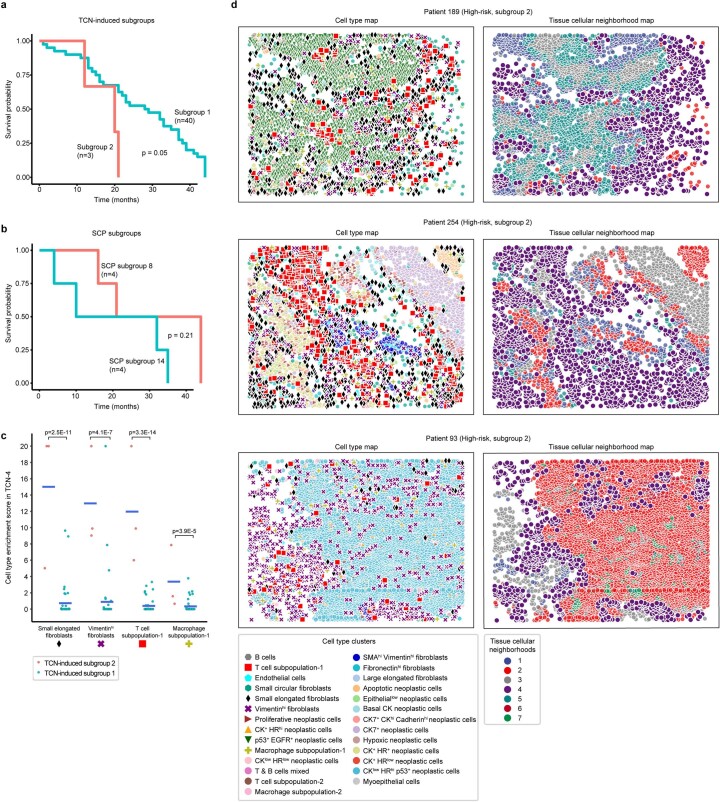
It is poorly understood how different cells in a tissue organize themselves to support tissue functions. We describe the CytoCommunity algorithm for the identification of tissue cellular neighborhoods (TCNs) based on cell phenotypes and their spatial distributions. CytoCommunity learns a mapping directly from the cell phenotype space to the TCN space using a graph neural network model without intermediate clustering of cell embeddings. By leveraging graph pooling, CytoCommunity enables de novo identification of condition-specific and predictive TCNs under the supervision of sample labels. Using several types of spatial omics data, we demonstrate that CytoCommunity can identify TCNs of variable sizes with substantial improvement over existing methods. By analyzing risk-stratified colorectal and breast cancer data, CytoCommunity revealed new granulocyte-enriched and cancer-associated fibroblast-enriched TCNs specific to high-risk tumors and altered interactions between neoplastic and immune or stromal cells within and between TCNs. CytoCommunity can perform unsupervised and supervised analyses of spatial omics maps and enable the discovery of condition-specific cell-cell communication patterns across spatial scales.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures








Similar articles
-
Cross-domain information fusion for enhanced cell population delineation in single-cell spatial-omics data.bioRxiv [Preprint]. 2024 May 14:2024.05.12.593710. doi: 10.1101/2024.05.12.593710. bioRxiv. 2024. PMID: 38798592 Free PMC article. Preprint.
-
Multi-granularity graph pooling for video-based person re-identification.Neural Netw. 2023 Mar;160:22-33. doi: 10.1016/j.neunet.2022.12.015. Epub 2022 Dec 28. Neural Netw. 2023. PMID: 36592527
-
Assembling spatial clustering framework for heterogeneous spatial transcriptomics data with GRAPHDeep.Bioinformatics. 2024 Jan 2;40(1):btae023. doi: 10.1093/bioinformatics/btae023. Bioinformatics. 2024. PMID: 38243703 Free PMC article.
-
Unsupervised and self-supervised deep learning approaches for biomedical text mining.Brief Bioinform. 2021 Mar 22;22(2):1592-1603. doi: 10.1093/bib/bbab016. Brief Bioinform. 2021. PMID: 33569575 Review.
-
Review of MR image segmentation techniques using pattern recognition.Med Phys. 1993 Jul-Aug;20(4):1033-48. doi: 10.1118/1.597000. Med Phys. 1993. PMID: 8413011 Review.
Cited by
-
Decoding Spatial Tissue Architecture: A Scalable Bayesian Topic Model for Multiplexed Imaging Analysis.bioRxiv [Preprint]. 2024 Oct 13:2024.10.08.617293. doi: 10.1101/2024.10.08.617293. bioRxiv. 2024. PMID: 39416145 Free PMC article. Preprint.
-
Next-generation spatial transcriptomics: unleashing the power to gear up translational oncology.MedComm (2020). 2024 Oct 6;5(10):e765. doi: 10.1002/mco2.765. eCollection 2024 Oct. MedComm (2020). 2024. PMID: 39376738 Free PMC article. Review.
-
Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics.Nat Rev Mol Cell Biol. 2024 Aug 21. doi: 10.1038/s41580-024-00768-2. Online ahead of print. Nat Rev Mol Cell Biol. 2024. PMID: 39169166 Review.
-
Multicell-Fold: geometric learning in folding multicellular life.ArXiv [Preprint]. 2024 Jul 22:arXiv:2407.07055v2. ArXiv. 2024. PMID: 39040638 Free PMC article. Preprint.
-
Cross-domain information fusion for enhanced cell population delineation in single-cell spatial-omics data.bioRxiv [Preprint]. 2024 May 14:2024.05.12.593710. doi: 10.1101/2024.05.12.593710. bioRxiv. 2024. PMID: 38798592 Free PMC article. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
