

Molecular and Cellular Mechanisms Affected in ALS
- PMID: 32854276
- PMCID: PMC7564998
- DOI: 10.3390/jpm10030101
Molecular and Cellular Mechanisms Affected in ALS
Abstract
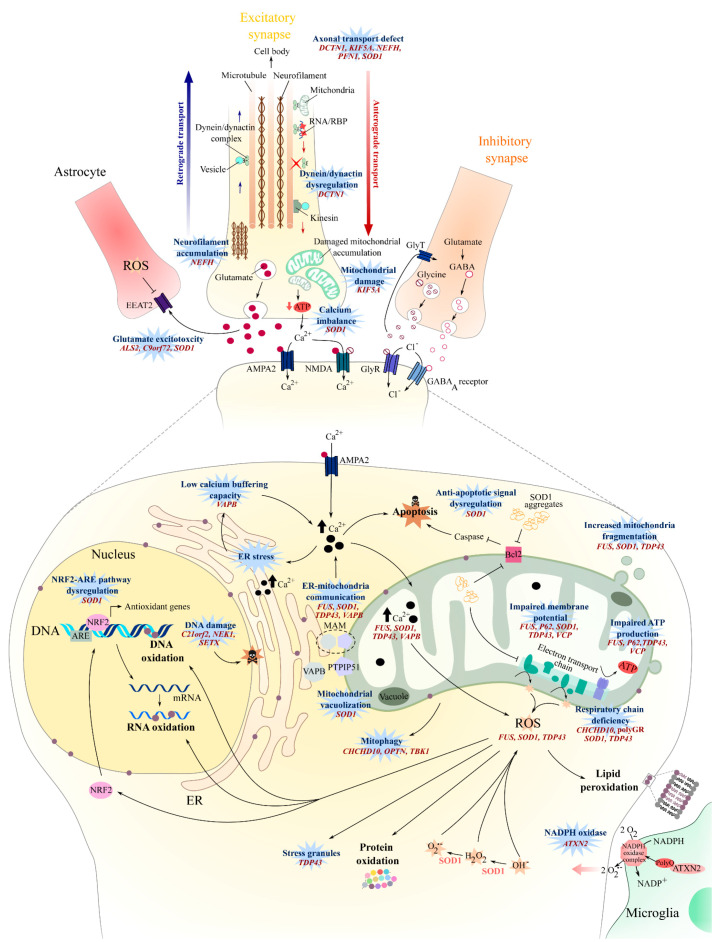
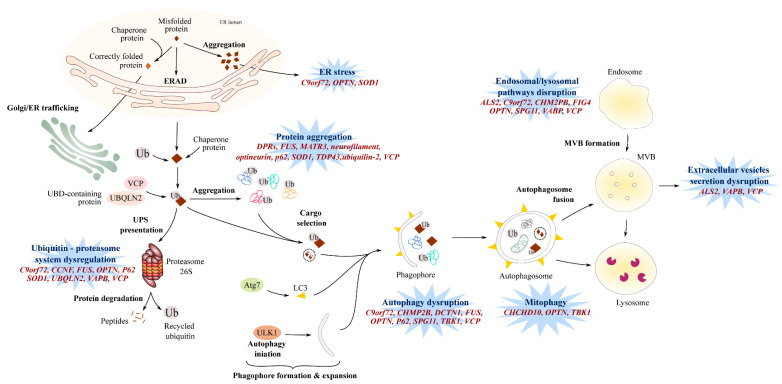
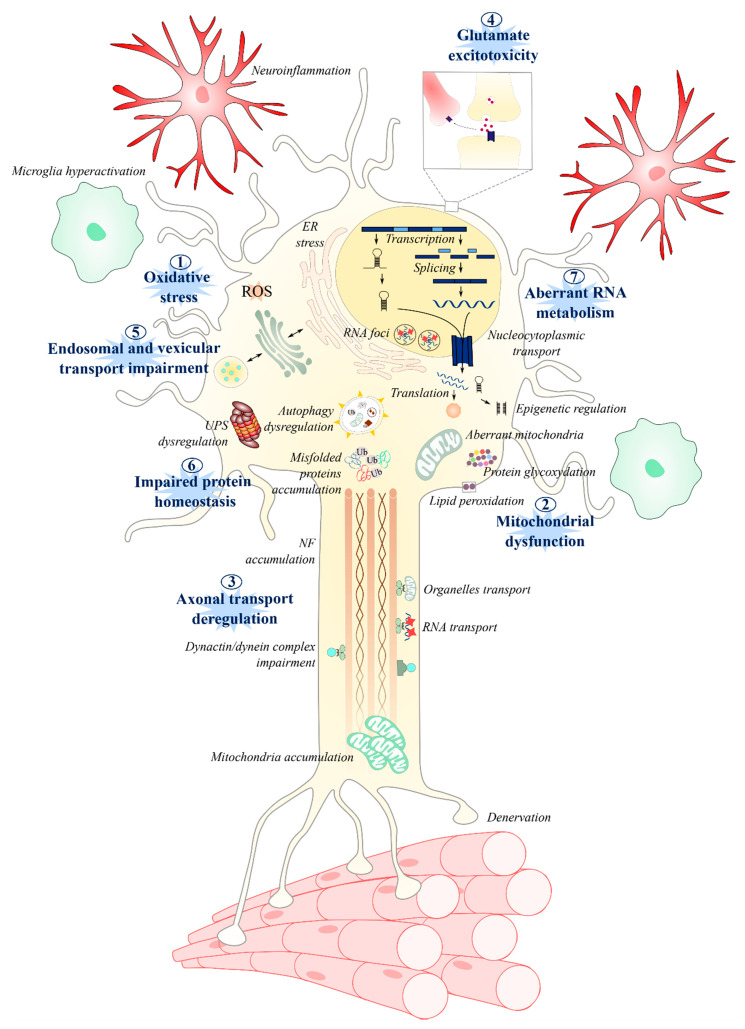
Amyotrophic lateral sclerosis (ALS) is a terminal late-onset condition characterized by the loss of upper and lower motor neurons. Mutations in more than 30 genes are associated to the disease, but these explain only ~20% of cases. The molecular functions of these genes implicate a wide range of cellular processes in ALS pathology, a cohesive understanding of which may provide clues to common molecular mechanisms across both familial (inherited) and sporadic cases and could be key to the development of effective therapeutic approaches. Here, the different pathways that have been investigated in ALS are summarized, discussing in detail: mitochondrial dysfunction, oxidative stress, axonal transport dysregulation, glutamate excitotoxicity, endosomal and vesicular transport impairment, impaired protein homeostasis, and aberrant RNA metabolism. This review considers the mechanistic roles of ALS-associated genes in pathology, viewed through the prism of shared molecular pathways.
Keywords: MND; RNA metabolism; autophagy; axonal transport; endocytosis; excitotoxicity; mitochondria dysfunction; oxidative stress; secretion.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Extracellular Vesicles in Amyotrophic Lateral Sclerosis.Life (Basel). 2022 Dec 31;13(1):121. doi: 10.3390/life13010121. Life (Basel). 2022. PMID: 36676070 Free PMC article. Review.
-
Genetics of amyotrophic lateral sclerosis: an update.Mol Neurodegener. 2013 Aug 13;8:28. doi: 10.1186/1750-1326-8-28. Mol Neurodegener. 2013. PMID: 23941283 Free PMC article. Review.
-
Mechanistic Insights of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: An Update on a Lasting Relationship.Metabolites. 2022 Mar 9;12(3):233. doi: 10.3390/metabo12030233. Metabolites. 2022. PMID: 35323676 Free PMC article. Review.
-
Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis.Front Neurosci. 2021 Dec 23;15:783624. doi: 10.3389/fnins.2021.783624. eCollection 2021. Front Neurosci. 2021. PMID: 35002606 Free PMC article. Review.
-
Mitochondrial dysfunction in amyotrophic lateral sclerosis.Biochim Biophys Acta. 2010 Jan;1802(1):45-51. doi: 10.1016/j.bbadis.2009.08.012. Epub 2009 Aug 26. Biochim Biophys Acta. 2010. PMID: 19715760 Free PMC article. Review.
Cited by
-
Extracellular Vesicles in Amyotrophic Lateral Sclerosis.Life (Basel). 2022 Dec 31;13(1):121. doi: 10.3390/life13010121. Life (Basel). 2022. PMID: 36676070 Free PMC article. Review.
-
Identification of Regulatory Factors and Prognostic Markers in Amyotrophic Lateral Sclerosis.Antioxidants (Basel). 2022 Feb 1;11(2):303. doi: 10.3390/antiox11020303. Antioxidants (Basel). 2022. PMID: 35204186 Free PMC article.
-
Discovery of Novel Inhibitors against ALS-Related SOD1(A4V) Aggregation through the Screening of a Chemical Library Using Differential Scanning Fluorimetry (DSF).Pharmaceuticals (Basel). 2024 Sep 27;17(10):1286. doi: 10.3390/ph17101286. Pharmaceuticals (Basel). 2024. PMID: 39458929 Free PMC article.
-
Status of ALS Treatment, Insights into Therapeutic Challenges and Dilemmas.J Pers Med. 2022 Sep 28;12(10):1601. doi: 10.3390/jpm12101601. J Pers Med. 2022. PMID: 36294741 Free PMC article. Review.
-
HGCA2.0: An RNA-Seq Based Webtool for Gene Coexpression Analysis in Homo sapiens.Cells. 2023 Jan 21;12(3):388. doi: 10.3390/cells12030388. Cells. 2023. PMID: 36766730 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
