

The Overlooked Fact: Fundamental Need for Spike-In Control for Virtually All Genome-Wide Analyses
- PMID: 26711261
- PMCID: PMC4760223
- DOI: 10.1128/MCB.00970-14
The Overlooked Fact: Fundamental Need for Spike-In Control for Virtually All Genome-Wide Analyses
Abstract
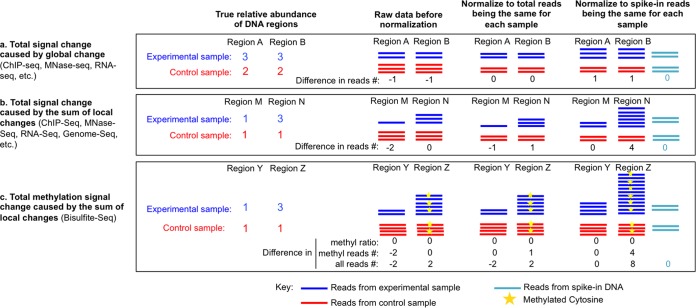
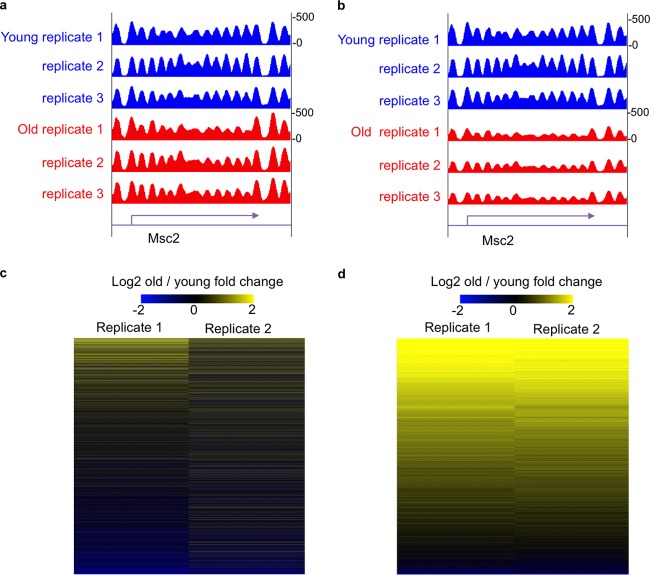
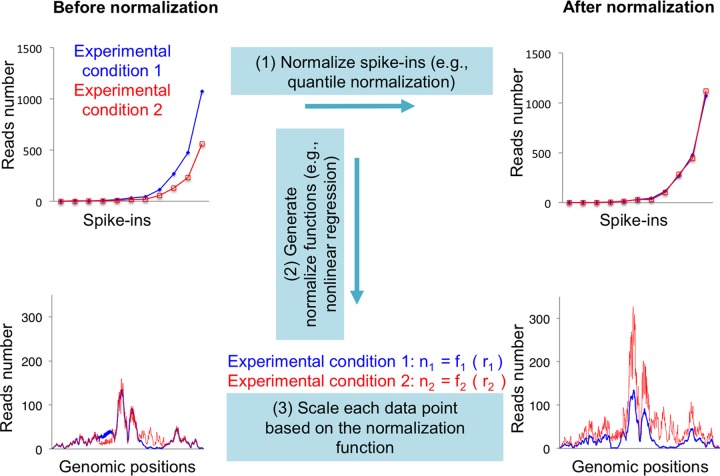
Genome-wide analyses of changes in gene expression, transcription factor occupancy on DNA, histone modification patterns on chromatin, genomic copy number variation, and nucleosome positioning have become popular in many modern laboratories, yielding a wealth of information during health and disease states. However, most of these studies have overlooked an inherent normalization problem that must be corrected with spike-in controls. Here we describe the reason why spike-in controls are so important and explain how to appropriately design and use spike-in controls for normalization. We also suggest ways to retrospectively renormalize data sets that were wrongly interpreted due to omission of spike-in controls.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Genome-wide mapping of nucleosome occupancy, histone modifications, and gene expression using next-generation sequencing technology.Methods Enzymol. 2012;513:297-313. doi: 10.1016/B978-0-12-391938-0.00013-6. Methods Enzymol. 2012. PMID: 22929775
-
ChIP on chip and ChIP-Seq assays: genome-wide analysis of transcription factor binding and histone modifications.Methods Mol Biol. 2015;1288:447-72. doi: 10.1007/978-1-4939-2474-5_26. Methods Mol Biol. 2015. PMID: 25827896
-
Massive parallel sequencing in animal genetics: wherefroms and wheretos.Anim Genet. 2010 Dec;41(6):561-9. doi: 10.1111/j.1365-2052.2010.02057.x. Anim Genet. 2010. PMID: 20477787 Review.
-
Mapping protein-DNA interactions using ChIP-sequencing.Methods Mol Biol. 2012;809:157-73. doi: 10.1007/978-1-61779-376-9_11. Methods Mol Biol. 2012. PMID: 22113275
-
Next generation sequencing technologies in cancer diagnostics and therapeutics: A mini review.Cell Mol Biol (Noisy-le-grand). 2015 Oct 30;61(5):91-102. Cell Mol Biol (Noisy-le-grand). 2015. PMID: 26522064 Review.
Cited by
-
Transcriptome view of a killer: African swine fever virus.Biochem Soc Trans. 2020 Aug 28;48(4):1569-1581. doi: 10.1042/BST20191108. Biochem Soc Trans. 2020. PMID: 32725217 Free PMC article. Review.
-
The Wild West of spike-in normalization.Nat Biotechnol. 2024 Sep;42(9):1343-1349. doi: 10.1038/s41587-024-02377-y. Nat Biotechnol. 2024. PMID: 39271835 No abstract available.
-
Optimization of an RNA-Seq Differential Gene Expression Analysis Depending on Biological Replicate Number and Library Size.Front Plant Sci. 2018 Feb 14;9:108. doi: 10.3389/fpls.2018.00108. eCollection 2018. Front Plant Sci. 2018. PMID: 29491871 Free PMC article.
-
The African Swine Fever Virus Transcriptome.J Virol. 2020 Apr 16;94(9):e00119-20. doi: 10.1128/JVI.00119-20. Print 2020 Apr 16. J Virol. 2020. PMID: 32075923 Free PMC article.
-
Chromatin structure-dependent histone incorporation revealed by a genome-wide deposition assay.Elife. 2021 May 10;10:e66290. doi: 10.7554/eLife.66290. Elife. 2021. PMID: 33970102 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources