

The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency
- PMID: 26152583
- PMCID: PMC4495168
- DOI: 10.1128/mBio.00465-15
The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency
Abstract
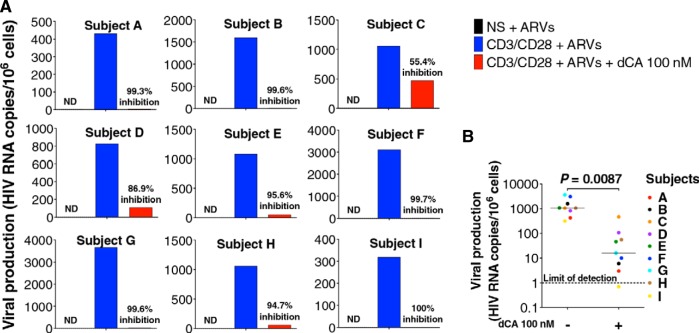
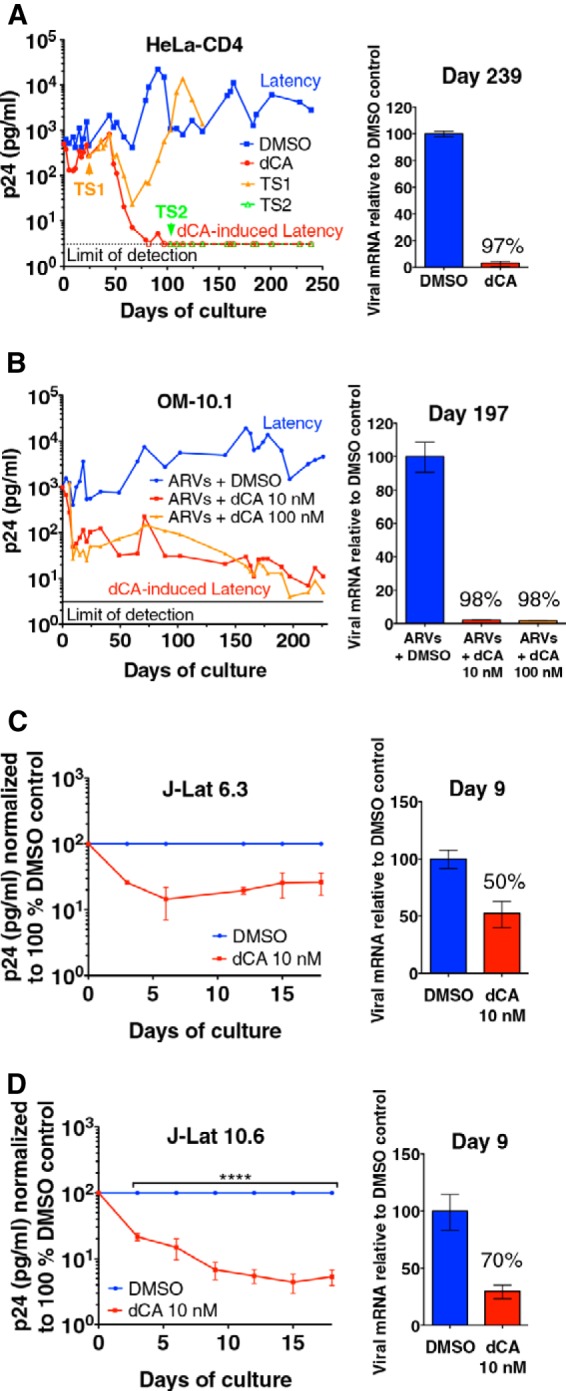
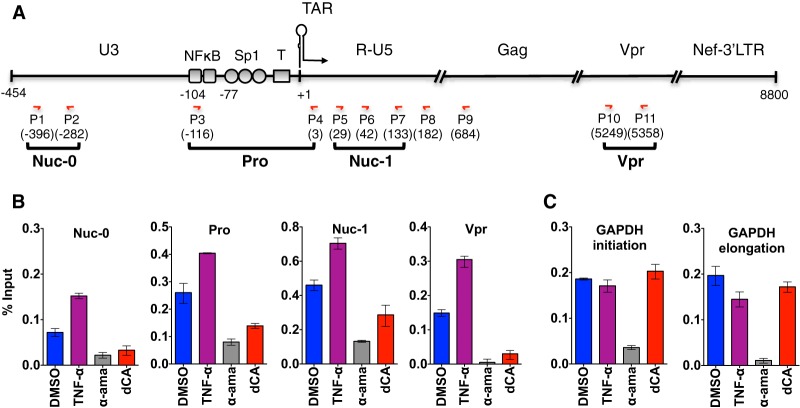
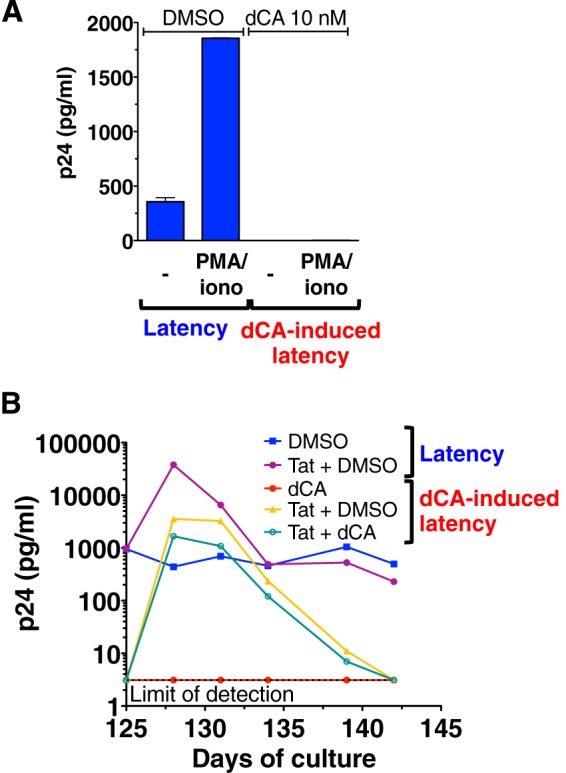
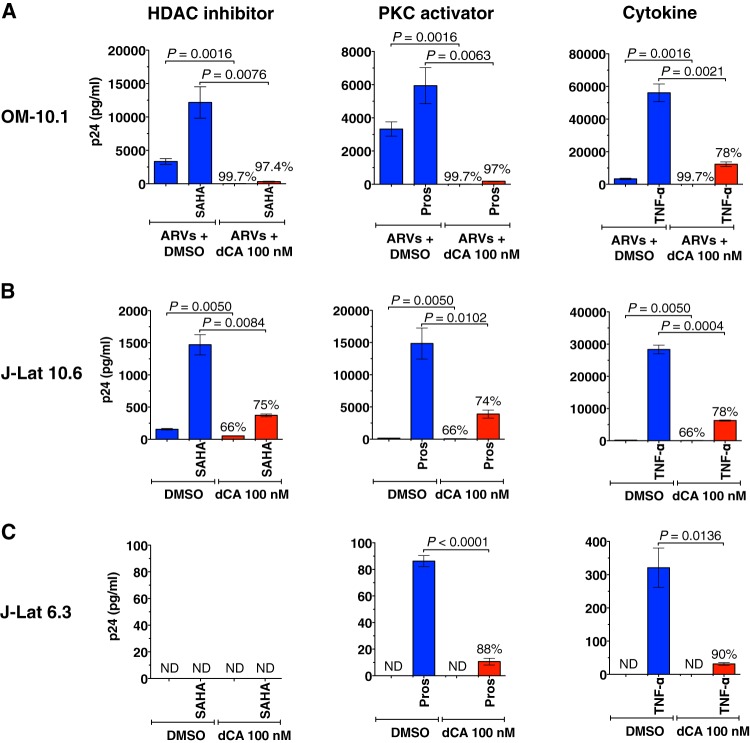
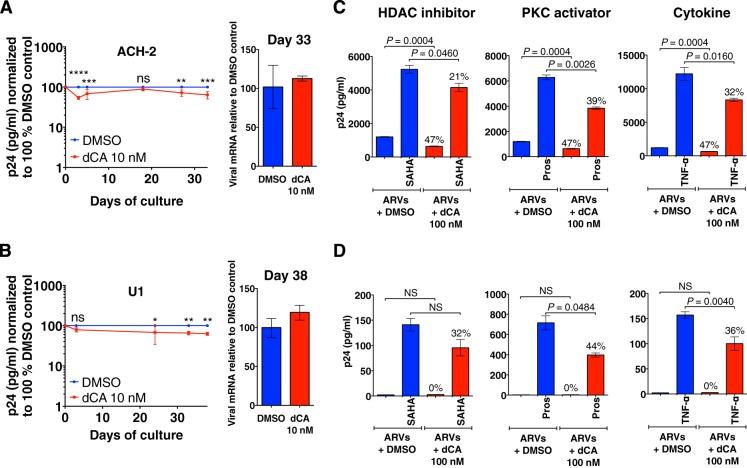
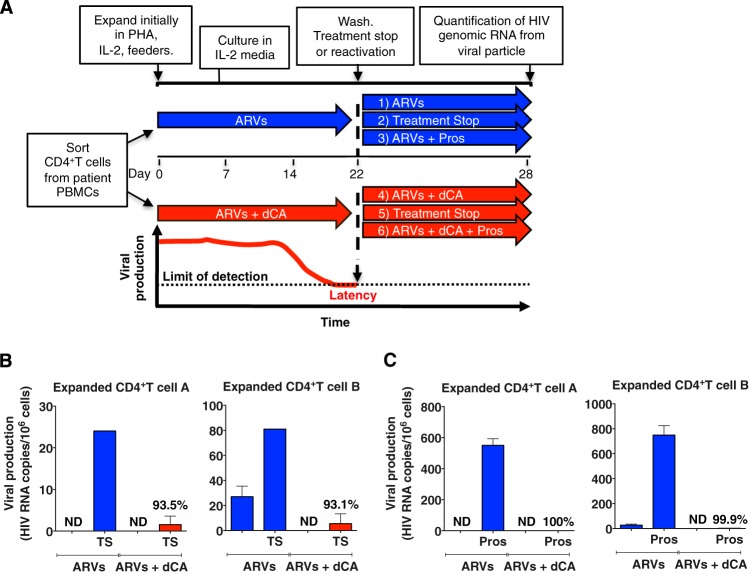
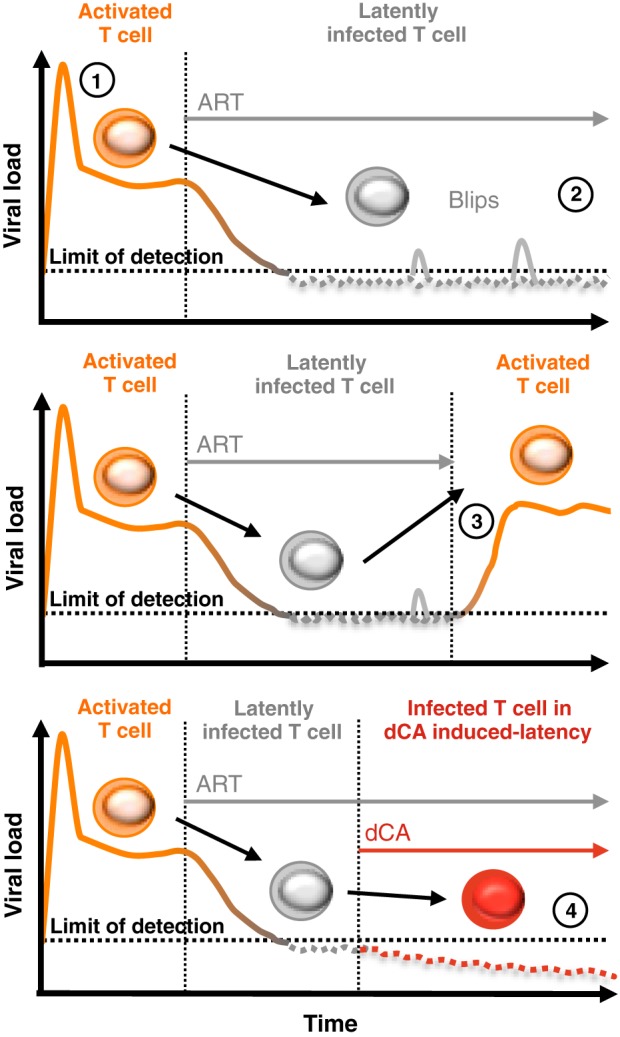
Antiretroviral therapy (ART) inhibits HIV-1 replication, but the virus persists in latently infected resting memory CD4(+) T cells susceptible to viral reactivation. The virus-encoded early gene product Tat activates transcription of the viral genome and promotes exponential viral production. Here we show that the Tat inhibitor didehydro-cortistatin A (dCA), unlike other antiretrovirals, reduces residual levels of viral transcription in several models of HIV latency, breaks the Tat-mediated transcriptional feedback loop, and establishes a nearly permanent state of latency, which greatly diminishes the capacity for virus reactivation. Importantly, treatment with dCA induces inactivation of viral transcription even after its removal, suggesting that the HIV promoter is epigenetically repressed. Critically, dCA inhibits viral reactivation upon CD3/CD28 or prostratin stimulation of latently infected CD4(+) T cells from HIV-infected subjects receiving suppressive ART. Our results suggest that inclusion of a Tat inhibitor in current ART regimens may contribute to a functional HIV-1 cure by reducing low-level viremia and preventing viral reactivation from latent reservoirs.
Importance: Antiretroviral therapy (ART) reduces HIV-1 replication to very low levels, but the virus persists in latently infected memory CD4(+) T cells, representing a long-lasting source of resurgent virus upon ART interruption. Based on the mode of action of didehydro-cortistatin A (dCA), a Tat-dependent transcription inhibitor, our work highlights an alternative approach to current HIV-1 eradication strategies to decrease the latent reservoir. In our model, dCA blocks the Tat feedback loop initiated after low-level basal reactivation, blocking transcriptional elongation and hence viral production from latently infected cells. Therefore, dCA combined with ART would be aimed at delaying or halting ongoing viral replication, reactivation, and replenishment of the latent viral reservoir. Thus, the latent pool of cells in an infected individual would be stabilized, and death of the long-lived infected memory T cells would result in a continuous decay of this pool over time, possibly culminating in the long-awaited sterilizing cure.
Copyright © 2015 Mousseau et al.
Figures
Similar articles
-
Didehydro-Cortistatin A: a new player in HIV-therapy?Expert Rev Anti Infect Ther. 2016;14(2):145-8. doi: 10.1586/14787210.2016.1122525. Epub 2015 Dec 11. Expert Rev Anti Infect Ther. 2016. PMID: 26581953 Free PMC article.
-
Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription.mBio. 2019 Jul 2;10(4):e01750-18. doi: 10.1128/mBio.01750-18. mBio. 2019. PMID: 31266880 Free PMC article.
-
Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat.mBio. 2019 Feb 5;10(1):e02662-18. doi: 10.1128/mBio.02662-18. mBio. 2019. PMID: 30723126 Free PMC article.
-
The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin A.J Infect Dis. 2021 Feb 15;223(12 Suppl 2):46-53. doi: 10.1093/infdis/jiaa681. J Infect Dis. 2021. PMID: 33586776 Free PMC article. Review.
-
Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure.Viruses. 2020 Apr 8;12(4):415. doi: 10.3390/v12040415. Viruses. 2020. PMID: 32276443 Free PMC article. Review.
Cited by
-
The multifaceted nature of HIV latency.J Clin Invest. 2020 Jul 1;130(7):3381-3390. doi: 10.1172/JCI136227. J Clin Invest. 2020. PMID: 32609095 Free PMC article. Review.
-
A Camptothetin Analog, Topotecan, Promotes HIV Latency via Interference with HIV Transcription and RNA Splicing.J Virol. 2023 Feb 28;97(2):e0163022. doi: 10.1128/jvi.01630-22. Epub 2023 Jan 31. J Virol. 2023. PMID: 36719238 Free PMC article.
-
HIV-1 transcriptional modulation: novel host factors and prospective therapeutic strategies.Curr Opin HIV AIDS. 2023 Sep 1;18(5):264-272. doi: 10.1097/COH.0000000000000808. Epub 2023 Jul 17. Curr Opin HIV AIDS. 2023. PMID: 37535041 Free PMC article. Review.
-
CDK8 inhibitors antagonize HIV-1 reactivation and promote provirus latency in T cells.J Virol. 2023 Sep 28;97(9):e0092323. doi: 10.1128/jvi.00923-23. Epub 2023 Sep 6. J Virol. 2023. PMID: 37671866 Free PMC article.
-
The BET bromodomain inhibitor apabetalone induces apoptosis of latent HIV-1 reservoir cells following viral reactivation.Acta Pharmacol Sin. 2019 Jan;40(1):98-110. doi: 10.1038/s41401-018-0027-5. Epub 2018 May 22. Acta Pharmacol Sin. 2019. PMID: 29789664 Free PMC article.
References
-
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi:10.1126/science.278.5341.1295. - DOI - PubMed
-
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi:10.1038/8394. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
