

Review
doi: 10.1038/nrg3788.
Epub 2014 Sep 16.
Identifying and mitigating bias in next-generation sequencing methods for chromatin biology
Affiliations
- PMID: 25223782
- PMCID: PMC4473780
- DOI: 10.1038/nrg3788
Item in Clipboard
Review
Identifying and mitigating bias in next-generation sequencing methods for chromatin biology
Nat Rev Genet.
2014 Nov.
Abstract
Next-generation sequencing (NGS) technologies have been used in diverse ways to investigate various aspects of chromatin biology by identifying genomic loci that are bound by transcription factors, occupied by nucleosomes or accessible to nuclease cleavage, or loci that physically interact with remote genomic loci. However, reaching sound biological conclusions from such NGS enrichment profiles requires many potential biases to be taken into account. In this Review, we discuss common ways in which biases may be introduced into NGS chromatin profiling data, approaches to diagnose these biases and analytical techniques to mitigate their effect.
Figures
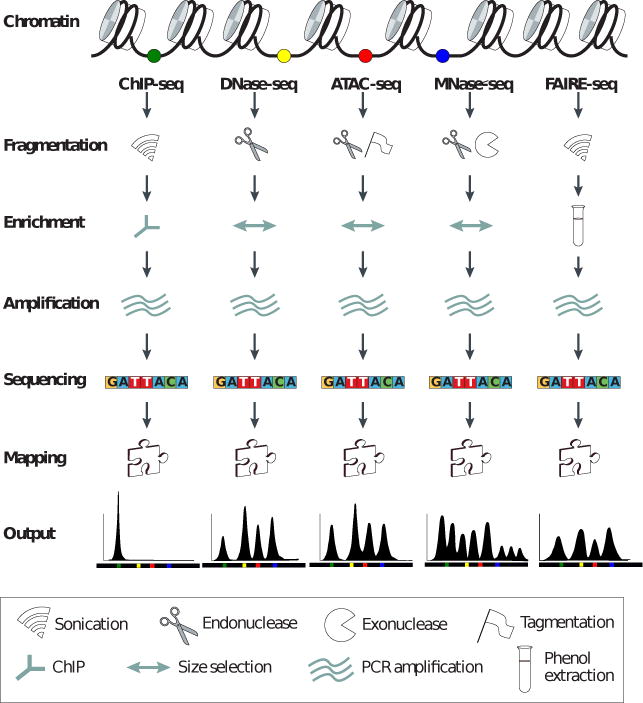
A genomic locus analyzed by complementary chromatin profiling experiments reveals different facets of chromatin structure; ChIP-seq reveals binding sites of specific transcription factors, DNase-seq and ATAC-seq reveal regions of open chromatin while MNase-seq identifies well-positioned nucleosomes. In ChIP-seq chromatin immunoprecipitation (ChIP) is used to extract DNA fragments that are bound to the target protein, either directly or via other proteins in a complex containing the target factor. In DNase-seq, chromatin is lightly digested by the DNase I endonuclease. Size selection is used to enrich for fragments that are produced in regions of chromatin where the DNA is highly sensitive to DNase I attack. ATAC-seq is an alternative to DNase-seq that uses an engineered Tn5 transposase to cleave DNA and to integrate primer DNA sequences into the cleaved genomic DNA. Micrococcal nuclease (MNase) is an endo-exo- nuclease that processively digests DNA until an obstruction such as a nucleosome is reached.

Chromatin structure, fragmentation, and enrichment, interact to produce biased patterns of enrichment across the genome. (a) Some transcription factors, such as CTCF, typically bind in short nucleosome-depleted regions that are flanked by arrays of nucleosomes. When carrying out DNase-seq short fragments are far more efficient than longer ones for identifying such sites. (b) Histones and other factors that associate with nucleosomes rather than linker regions may also be located in DNase I hypersensitive regions. Longer fragments may be more efficient for detecting the binding of such factors. (c) Some factors bind in linker regions that are flanked by loosely unorganized nucleosomes. Such regions can be enriched in both long and short fragments in DNase-seq. (d) In ChIP-seq chromatin is typically fragmented by sonication. Like DNase-seq sonication is more efficient in regions of open chromatin. Factors bound in open chromatin contexts are more likely to be identified by ChIP-seq.
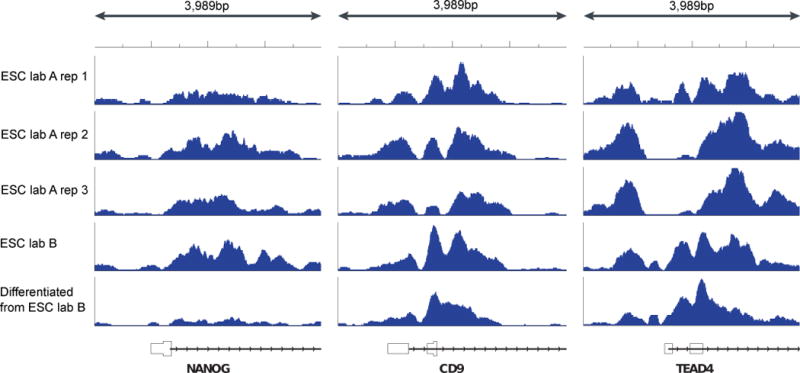
Several factors including fragmentation, immunoprecipitation conditions, and PCR biases can lead to different patterns of H3K4me3 enrichment at gene promoters in the same cell line. Coarse characteristics of H3K4me3 enrichment are consistent between samples, for example the depletion of H3K4me3 immediately upstream of the transcription start site of a core set of genes. Closer inspection reveals clear qualitative and quantitative differences between samples. For example, some samples show sharper peaks, perhaps due to differences in MNase digestion conditions and fragment selection. Regions that appear to be different between ES cells and differentiated cells in ChIP-seq samples produced by laboratory B also show variability in ES cell ChIP-seq replicates produced by laboratory A. These differences cannot be eliminated simply by scaling read counts to account for differences in read depth as the effects are not uniform across all genes. Quantitative comparisons of ChIP-seq signal are problematic unless biological replicates are done and protocols are carried out in a highly consistent way to produce data with comparable characteristics. Modeling bias can help reduce the amount of unexplained variability and increase sensitivity in detecting true differences between sample groups.
Similar articles
-
ChIP-Seq: Library Preparation and Sequencing.Methods Mol Biol. 2016;1402:101-117. doi: 10.1007/978-1-4939-3378-5_9. Methods Mol Biol. 2016. PMID: 26721486
-
Unified Analysis of Multiple ChIP-Seq Datasets.Methods Mol Biol. 2021;2198:451-465. doi: 10.1007/978-1-0716-0876-0_33. Methods Mol Biol. 2021. PMID: 32822050
-
Next-generation sequencing applied to flower development: ChIP-Seq.Methods Mol Biol. 2014;1110:413-29. doi: 10.1007/978-1-4614-9408-9_24. Methods Mol Biol. 2014. PMID: 24395273
-
Tn5 Transposase Applied in Genomics Research.Int J Mol Sci. 2020 Nov 6;21(21):8329. doi: 10.3390/ijms21218329. Int J Mol Sci. 2020. PMID: 33172005 Free PMC article. Review.
-
Library preparation methods for next-generation sequencing: tone down the bias.Exp Cell Res. 2014 Mar 10;322(1):12-20. doi: 10.1016/j.yexcr.2014.01.008. Epub 2014 Jan 15. Exp Cell Res. 2014. PMID: 24440557 Review.
Cited by
-
Unraveling the 3D genome: genomics tools for multiscale exploration.Trends Genet. 2015 Jul;31(7):357-72. doi: 10.1016/j.tig.2015.03.010. Epub 2015 Apr 14. Trends Genet. 2015. PMID: 25887733 Free PMC article. Review.
-
Paired-end mappability of transposable elements in the human genome.Mob DNA. 2019 Jul 10;10:29. doi: 10.1186/s13100-019-0172-5. eCollection 2019. Mob DNA. 2019. PMID: 31320939 Free PMC article.
-
An ATAC-seq atlas of chromatin accessibility in mouse tissues.Sci Data. 2019 May 20;6(1):65. doi: 10.1038/s41597-019-0071-0. Sci Data. 2019. PMID: 31110271 Free PMC article.
-
Integrating ChIP-seq with other functional genomics data.Brief Funct Genomics. 2018 Mar 1;17(2):104-115. doi: 10.1093/bfgp/ely002. Brief Funct Genomics. 2018. PMID: 29579165 Free PMC article. Review.
-
BIDCHIPS: bias decomposition and removal from ChIP-seq data clarifies true binding signal and its functional correlates.Epigenetics Chromatin. 2015 Sep 17;8:33. doi: 10.1186/s13072-015-0028-2. eCollection 2015. Epigenetics Chromatin. 2015. PMID: 26388941 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
