

CCDC22 deficiency in humans blunts activation of proinflammatory NF-κB signaling
- PMID: 23563313
- PMCID: PMC3635737
- DOI: 10.1172/JCI66466
CCDC22 deficiency in humans blunts activation of proinflammatory NF-κB signaling
Abstract
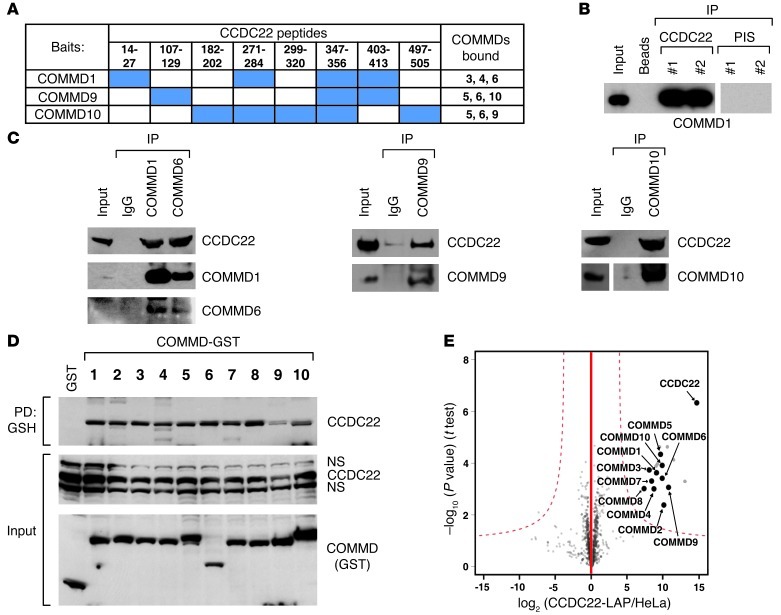
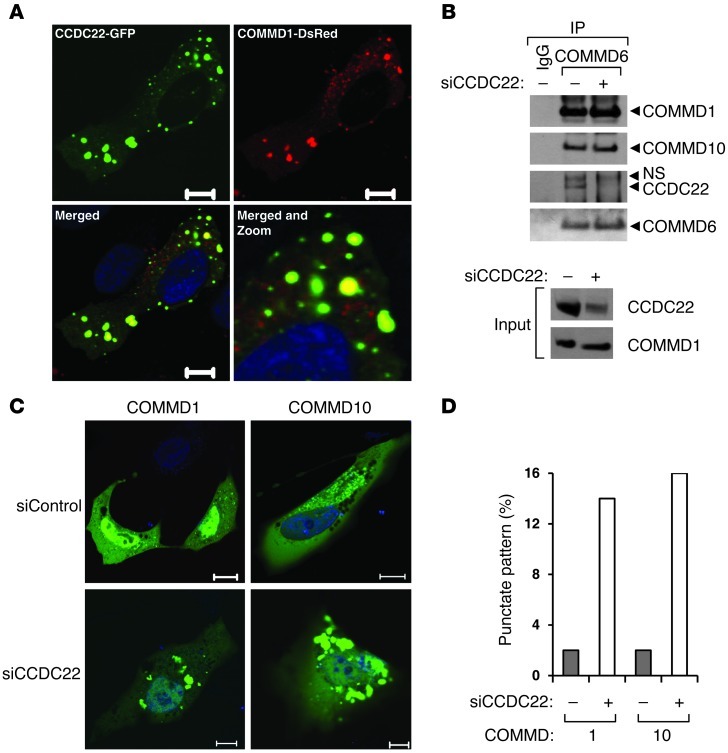
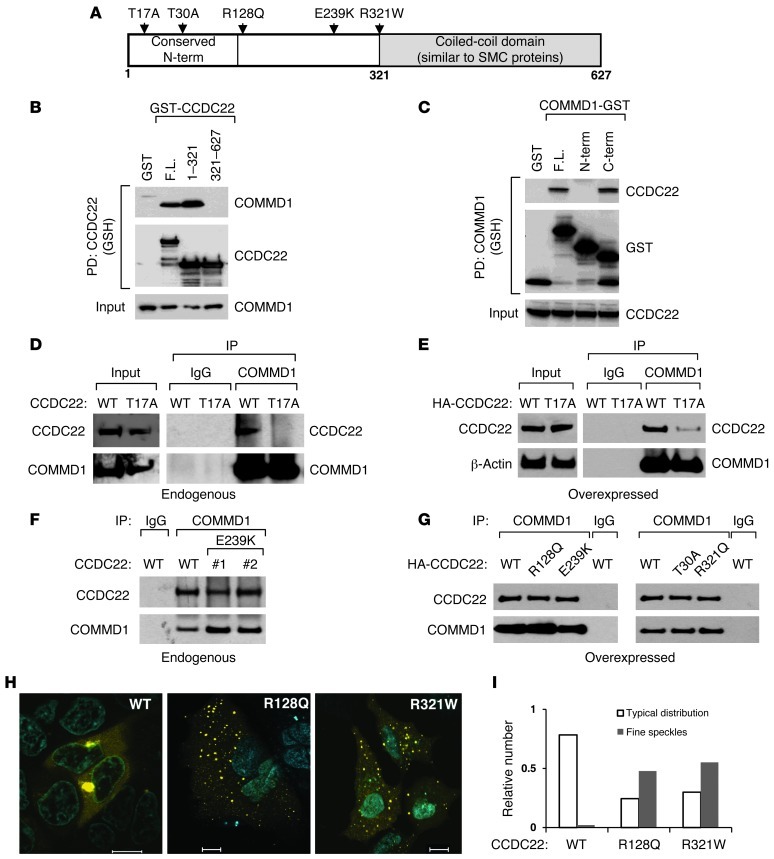
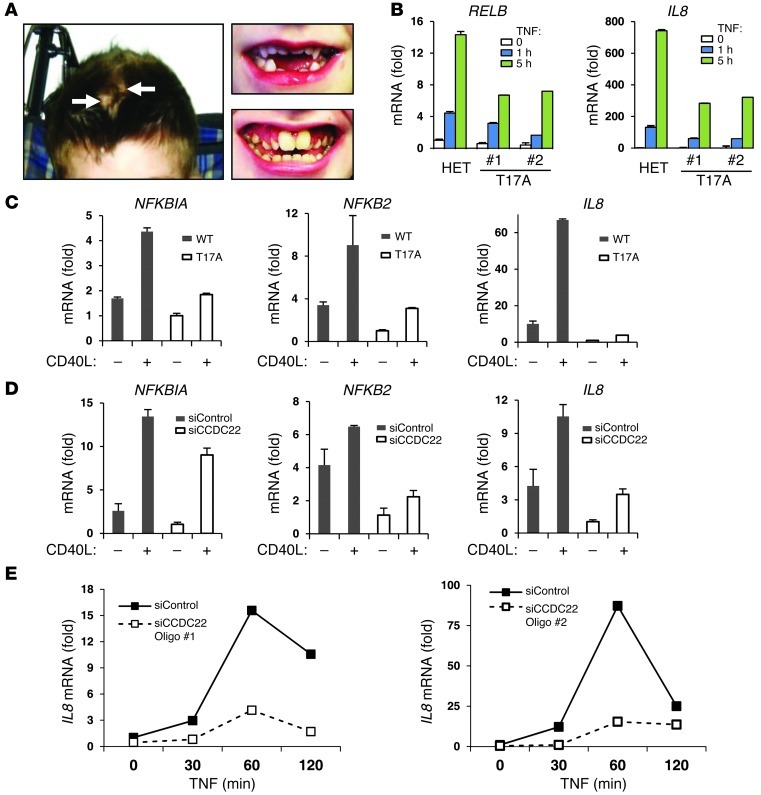
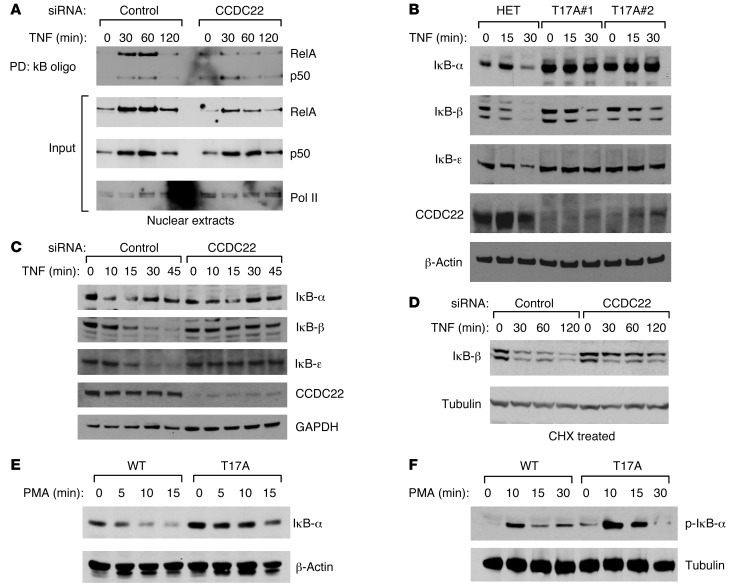
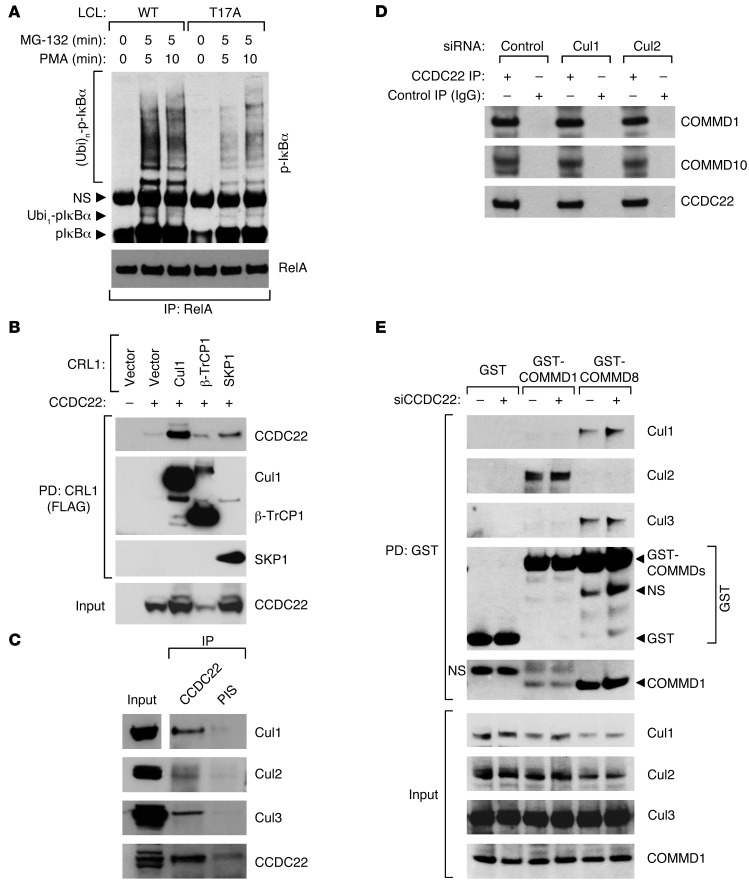
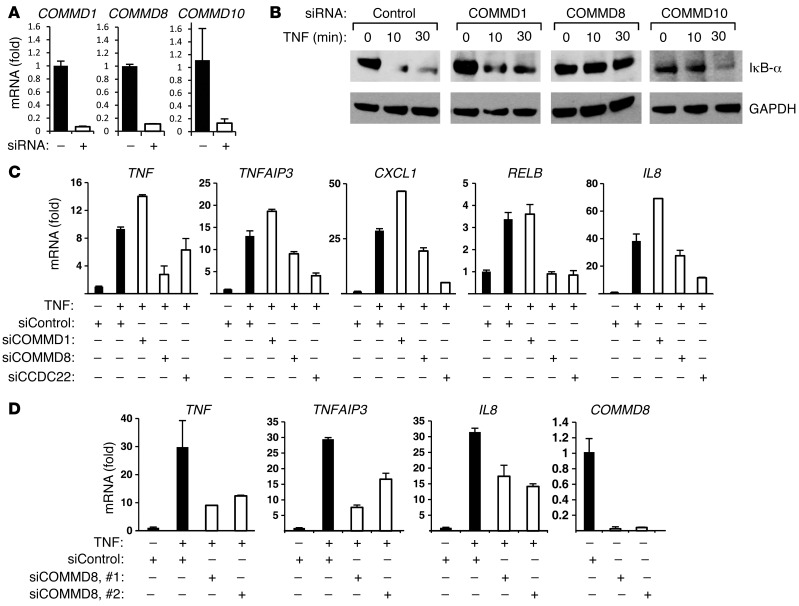
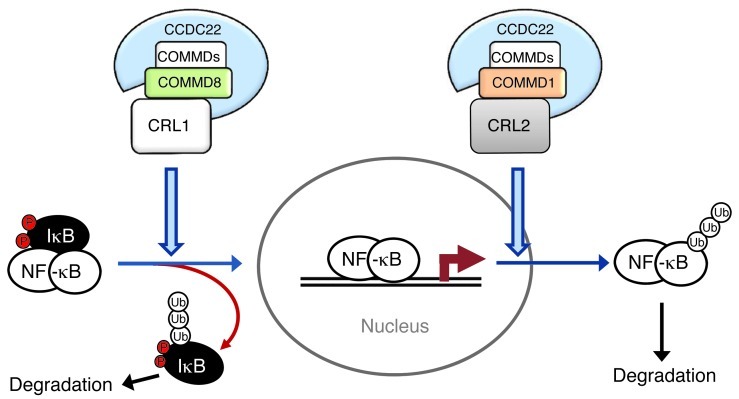
NF-κB is a master regulator of inflammation and has been implicated in the pathogenesis of immune disorders and cancer. Its regulation involves a variety of steps, including the controlled degradation of inhibitory IκB proteins. In addition, the inactivation of DNA-bound NF-κB is essential for its regulation. This step requires a factor known as copper metabolism Murr1 domain-containing 1 (COMMD1), the prototype member of a conserved gene family. While COMMD proteins have been linked to the ubiquitination pathway, little else is known about other family members. Here we demonstrate that all COMMD proteins bind to CCDC22, a factor recently implicated in X-linked intellectual disability (XLID). We showed that an XLID-associated CCDC22 mutation decreased CCDC22 protein expression and impaired its binding to COMMD proteins. Moreover, some affected individuals displayed ectodermal dysplasia, a congenital condition that can result from developmental NF-κB blockade. Indeed, patient-derived cells demonstrated impaired NF-κB activation due to decreased IκB ubiquitination and degradation. In addition, we found that COMMD8 acted in conjunction with CCDC22 to direct the degradation of IκB proteins. Taken together, our results indicate that CCDC22 participates in NF-κB activation and that its deficiency leads to decreased IκB turnover in humans, highlighting an important regulatory component of this pathway.
Figures
Similar articles
-
Characterization of COMMD protein-protein interactions in NF-kappaB signalling.Biochem J. 2006 Aug 15;398(1):63-71. doi: 10.1042/BJ20051664. Biochem J. 2006. PMID: 16573520 Free PMC article.
-
Ubiquitin-associated domain-containing ubiquitin regulatory X (UBX) protein UBXN1 is a negative regulator of nuclear factor κB (NF-κB) signaling.J Biol Chem. 2015 Apr 17;290(16):10395-405. doi: 10.1074/jbc.M114.631689. Epub 2015 Feb 13. J Biol Chem. 2015. PMID: 25681446 Free PMC article.
-
The COMMD Family Regulates Plasma LDL Levels and Attenuates Atherosclerosis Through Stabilizing the CCC Complex in Endosomal LDLR Trafficking.Circ Res. 2018 Jun 8;122(12):1648-1660. doi: 10.1161/CIRCRESAHA.117.312004. Epub 2018 Mar 15. Circ Res. 2018. PMID: 29545368
-
Tuning NF-κB activity: a touch of COMMD proteins.Biochim Biophys Acta. 2013 Dec;1832(12):2315-21. doi: 10.1016/j.bbadis.2013.09.014. Epub 2013 Sep 29. Biochim Biophys Acta. 2013. PMID: 24080195 Review.
-
Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity.Annu Rev Immunol. 2000;18:621-63. doi: 10.1146/annurev.immunol.18.1.621. Annu Rev Immunol. 2000. PMID: 10837071 Review.
Cited by
-
Structural Organization of the Retriever-CCC Endosomal Recycling Complex.Res Sq [Preprint]. 2023 Jun 16:rs.3.rs-3026818. doi: 10.21203/rs.3.rs-3026818/v1. Res Sq. 2023. Update in: Nat Struct Mol Biol. 2024 Jun;31(6):910-924. doi: 10.1038/s41594-023-01184-4 PMID: 37397996 Free PMC article. Updated. Preprint.
-
Canonical and Divergent N-Terminal HBx Isoform Proteins Unveiled: Characteristics and Roles during HBV Replication.Biomedicines. 2021 Nov 16;9(11):1701. doi: 10.3390/biomedicines9111701. Biomedicines. 2021. PMID: 34829930 Free PMC article.
-
Proteomic analysis and identification of cellular interactors of the giant ubiquitin ligase HERC2.J Proteome Res. 2015 Feb 6;14(2):953-66. doi: 10.1021/pr501005v. Epub 2014 Dec 15. J Proteome Res. 2015. PMID: 25476789 Free PMC article.
-
Missense variant in CCDC22 causes X-linked recessive intellectual disability with features of Ritscher-Schinzel/3C syndrome.Eur J Hum Genet. 2015 May;23(5):633-8. doi: 10.1038/ejhg.2014.109. Epub 2014 Jun 11. Eur J Hum Genet. 2015. PMID: 24916641 Free PMC article.
-
X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes.Mol Psychiatry. 2016 Jan;21(1):133-48. doi: 10.1038/mp.2014.193. Epub 2015 Feb 3. Mol Psychiatry. 2016. PMID: 25644381 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
