

Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling
- PMID: 19052195
- PMCID: PMC2614130
- DOI: 10.1523/JNEUROSCI.4407-08.2008
Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling
Abstract
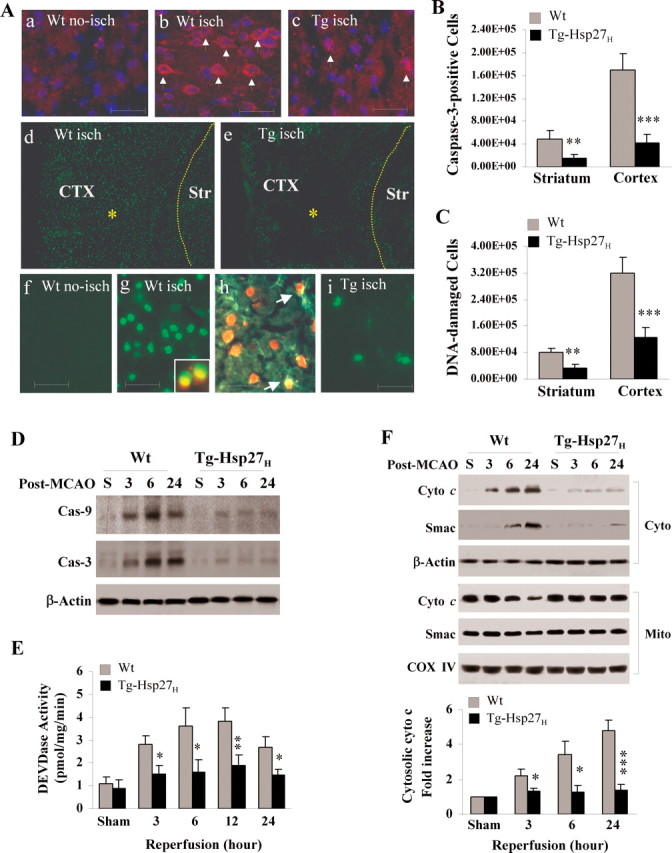
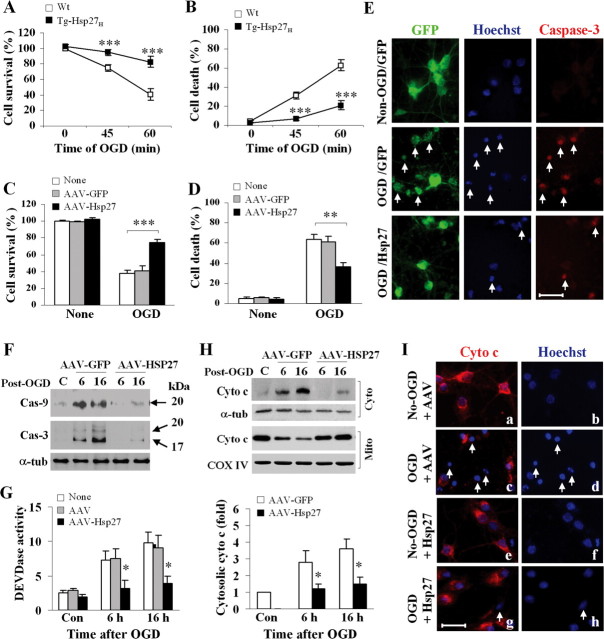
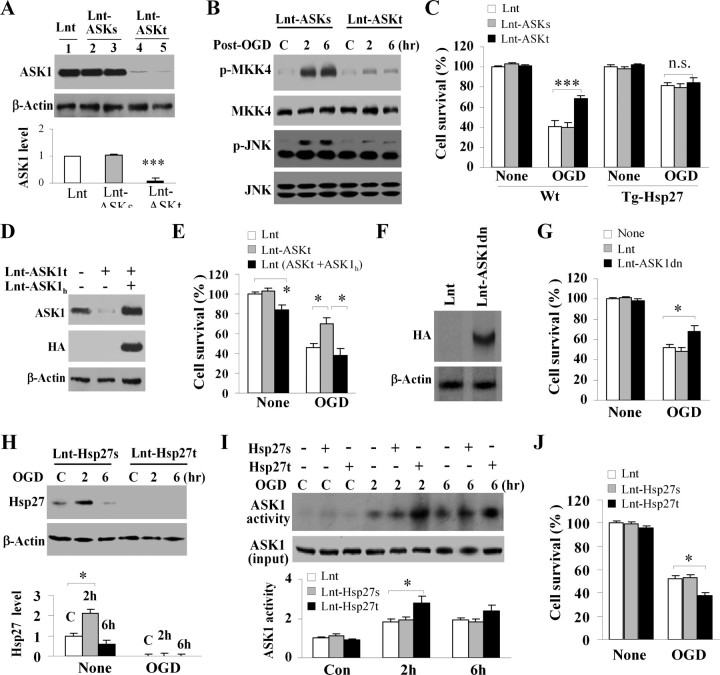
Heat shock protein 27 (Hsp27), a recently discovered member of the heat shock protein family, is markedly induced in the brain after cerebral ischemia and other injury states. In non-neuronal systems, Hsp27 has potent cell death-suppressing functions. However, the mechanism of Hsp27-mediated neuroprotection has not yet been elucidated. Using transgenic and viral overexpression of Hsp27, we investigated the molecular mechanism by which Hsp27 exerts its neuroprotective effect. Overexpression of Hsp27 conferred long-lasting tissue preservation and neurobehavioral recovery, as measured by infarct volume, sensorimotor function, and cognitive tasks up to 3 weeks following focal cerebral ischemia. Examination of signaling pathways critical to neuronal death demonstrated that Hsp27 overexpression led to the suppression of the MKK4/JNK kinase cascade. While Hsp27 overexpression did not suppress activation of an upstream regulatory kinase of the MKK/JNK cascade, ASK1, Hsp27 effectively inhibited ASK1 activity via a physical association through its N-terminal domain and the kinase domain of ASK1. The N-terminal region of Hsp27 was required for neuroprotective function against in vitro ischemia. Moreover, knockdown of ASK1 or inhibition of the ASK1/MKK4 cascade effectively inhibited cell death following neuronal ischemia. This underscores the importance of this kinase cascade in the progression of ischemic neuronal death. Inhibition of PI3K had no effect on Hsp27-mediated neuroprotection, suggesting that Hsp27 does not promote cell survival via activation of PI3K/Akt. Based on these findings, we conclude that overexpression of Hsp27 confers long-lasting neuroprotection against ischemic brain injury via a previously unexplored association and inhibition of ASK1 kinase signaling.
Figures







Similar articles
-
Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway.Antioxid Redox Signal. 2012 Sep 1;17(5):719-32. doi: 10.1089/ars.2011.4298. Epub 2012 May 14. Antioxid Redox Signal. 2012. PMID: 22356734 Free PMC article.
-
Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury.J Neurosci. 2012 Feb 22;32(8):2667-82. doi: 10.1523/JNEUROSCI.5169-11.2012. J Neurosci. 2012. PMID: 22357851 Free PMC article.
-
Human-derived physiological heat shock protein 27 complex protects brain after focal cerebral ischemia in mice.PLoS One. 2013 Jun 13;8(6):e66001. doi: 10.1371/journal.pone.0066001. Print 2013. PLoS One. 2013. PMID: 23785464 Free PMC article.
-
Heat shock proteins in the brain: role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential.Transl Stroke Res. 2013 Dec;4(6):685-92. doi: 10.1007/s12975-013-0271-4. Epub 2013 Aug 3. Transl Stroke Res. 2013. PMID: 24323422 Free PMC article. Review.
-
Heat shock protein 27 as a neuroprotective biomarker and a suitable target for stem cell therapy and pharmacotherapy in ischemic stroke.Cell Biol Int. 2020 Feb;44(2):356-367. doi: 10.1002/cbin.11237. Epub 2019 Sep 23. Cell Biol Int. 2020. PMID: 31502740 Review.
Cited by
-
Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway.Antioxid Redox Signal. 2012 Sep 1;17(5):719-32. doi: 10.1089/ars.2011.4298. Epub 2012 May 14. Antioxid Redox Signal. 2012. PMID: 22356734 Free PMC article.
-
Heat shock protein B1-deficient mice display impaired wound healing.PLoS One. 2013 Oct 15;8(10):e77383. doi: 10.1371/journal.pone.0077383. eCollection 2013. PLoS One. 2013. PMID: 24143227 Free PMC article.
-
High-salt diet downregulates TREM2 expression and blunts efferocytosis of macrophages after acute ischemic stroke.J Neuroinflammation. 2021 Apr 12;18(1):90. doi: 10.1186/s12974-021-02144-9. J Neuroinflammation. 2021. PMID: 33845849 Free PMC article.
-
Transgenic overexpression of 14-3-3 zeta protects hippocampus against endoplasmic reticulum stress and status epilepticus in vivo.PLoS One. 2013;8(1):e54491. doi: 10.1371/journal.pone.0054491. Epub 2013 Jan 24. PLoS One. 2013. PMID: 23359526 Free PMC article.
-
Oxidative stress markers in the brain of patients with cirrhosis and hepatic encephalopathy.Hepatology. 2010 Jul;52(1):256-65. doi: 10.1002/hep.23656. Hepatology. 2010. PMID: 20583283 Free PMC article.
References
-
- An JJ, Lee YP, Kim SY, Lee SH, Lee MJ, Jeong MS, Kim DW, Jang SH, Yoo KY, Won MH, Kang TC, Kwon OS, Cho SW, Lee KS, Park J, Eum WS, Choi SY. Transduced human PEP-1-heat shock protein 27 efficiently protects against brain ischemic insult. FEBS J. 2008;275:1296–1308. - PubMed
-
- Arya R, Mallik M, Lakhotia SC. Heat shock genes—integrating cell survival and death. J Biosci. 2007;32:595–610. - PubMed
-
- Badin RA, Lythgoe MF, van der Weerd L, Thomas DL, Gadian DG, Latchman DS. Neuroprotective effects of virally delivered HSPs in experimental stroke. J Cereb Blood Flow Metab. 2006;26:371–381. - PubMed
-
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 NS056118-02/NS/NINDS NIH HHS/United States
- R01 NS036736-10/NS/NINDS NIH HHS/United States
- R01 NS056118/NS/NINDS NIH HHS/United States
- R01 NS045048-05/NS/NINDS NIH HHS/United States
- R01 NS043802/NS/NINDS NIH HHS/United States
- R01 NS043802-05/NS/NINDS NIH HHS/United States
- NS36736/NS/NINDS NIH HHS/United States
- R01 NS056118-01/NS/NINDS NIH HHS/United States
- NS43802/NS/NINDS NIH HHS/United States
- NS 56118/NS/NINDS NIH HHS/United States
- R01 NS056118-03/NS/NINDS NIH HHS/United States
- NS45048/NS/NINDS NIH HHS/United States
- R01 NS036736/NS/NINDS NIH HHS/United States
- R01 NS045048/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous