

SUMMARY
Humans with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), a T cell–driven autoimmune disease caused by impaired central tolerance, are susceptible to developing chronic fungal infection and esophageal squamous cell carcinoma (ESCC). However, the relationship between autoreactive T cells and chronic fungal infection in ESCC development remains unclear. We find that kinase-dead Ikkα knockin mice develop phenotypes reminiscent of APECED, including impaired central tolerance, autoreactive T cells, chronic fungal infection, and ESCCs expressing specific human ESCC markers. Using this model, we investigated the potential link between ESCC and fungal infection. Autoreactive CD4 T cells permit fungal infection and incite tissue injury and inflammation. Antifungal treatment or depletion of autoreactive CD4 T cells rescues, whereas oral fungal administration promotes, ESCC development. Inhibition of inflammation or EGFR activity decreases fungal burden. Importantly, fungal infection is highly associated with ESCCs in non-autoimmune human patients. Therefore, autoreactive T cells and chronic fungal infection, fostered by inflammation and epithelial injury, promote ESCC development.
Keywords: IKKalpha in immunity and carcinogenesis, fungal infection, autoimmune disease, central tolerance, autoreactive T cells, squamous cell carcinoma
INTRODUCTION
A causal relationship between T cell–mediated autoimmunity and fungal infection in carcinogenesis has not been appreciated. Patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), a T cell–driven autoimmune disease caused by impaired central tolerance, are susceptible to developing chronic fungal infection and esophageal squamous cell carcinoma (ESCC) (Manley et al., 2011; Mathis and Benoist, 2009; Rautemaa et al., 2007). The defective central tolerance in APECED patients is due to mutations in the autoimmune regulator (AIRE) gene (Mathis and Benoist, 2009). Although the T cells from APECED patients have been suggested to favor fungal growth (Rautemaa et al., 2007), it is still unknown whether the autoreactive T cells permit chronic fungal infection, which in turn drives ESCC development.
Central tolerance is established in the thymus, where self-reactive T cells are deleted by interacting with medullary thymic epithelial cells (mTECs) that express tissue-restricted antigens (TRAs). AIRE, a transcriptional factor, regulates the expression of TRAs, cytokines, and chemokines in mTECs, which determine the thymic microenvironment required for selecting proper T-cell repertoires (Akiyama et al., 2005). Classical helper (CD4) and cytotoxic (CD8) T cells express αβ T cell receptors (TCRs) that use random Variable (V), Diversity (D), and Joining (J) gene rearrangement to generate enormously diverse repertoire including many autoreactive specificities. A lack of mTECs allows the autoreactive T cells to evade the negative selection in the thymus and move to the periphery. These autoreactive T cells incite tissue injury and provoke systemic inflammation. Alterations in TCR Vβ loci and increased Vβ5.1 repertoire have been reported in APECED patients (Kogawa et al., 2002; Niemi et al., 2015). Notably, Aire−/− mice only recapitulate part of phenotypes of APECED (Anderson et al., 2002; Ramsey et al., 2002). NF-kB regulates Aire expression and mTEC development (Akiyama et al., 2005; Akiyama et al., 2008). Mice deficient in genes encoding NF-kB molecules exhibit impaired central tolerance-induced autoimmune diseases, but none of these mice display fungal infection–associated tumorigenesis. Now, there are no suitable tools for studying the relationship between autoreactive T cells and fungal infection.
The fungi kingdom includes a vast and highly diverse array of species that are ubiquitous in the environment (Underhill and Iliev, 2014). Innate and adaptive T-helper cells provide essential protection against fungal infection and prevent the expansion of fungi in the human body. The esophagus, one of the gastrointestinal (GI) organs, is covered by a mucosal squamous epithelial layer and frequently has contact with various infectious agents from the environment through the mouth. Fungating, ulcerating, and esophagitis are frequently observed in human ESCC (HESCC), one of the deadliest cancers (Stoner and Gupta, 2001), indicating a close association between environmental fungi and HESCC. To date, the etiological causes of HESCCs remain unclear.
Squamous epithelial cells build up the stratified or pseudostratified layers that cover the surface of the skin, lungs, esophagus, oral cavity, and nasopharynx, and shield these organs from environmental contacts. IKKα is essential for the formation of the epidermis and the maintenance of skin homeostasis (Hu et al., 1999; Liu et al., 2008). IKKα functions as a tumor suppressor in the skin: its somatic ablation in keratinocytes expands epidermal-basal keratinocytes expressing keratin 5/14 (K5/14) and induces spontaneous skin SCC (Hu et al., 1999; Hu et al., 2001; Liu et al., 2008; Xia et al., 2010). IKKα deletion elevates EGFR activity by upregulating the transcription of Egf, HB-Egf, and a disintegrin and metalloproteinase domain (Adam) genes (Liu et al., 2008). Inactivation of EGFR prevents IKKα deletion-induced skin tumorigenesis. Also, IKKα deletion promotes cell cycle progression and genomic instability, thereby accelerating skin tumor progression (Xia et al., 2013). Furthermore, IKKα reduction enhances ΔNp63 and Trim29 but decreases p53 and Rb expression, resulting in the formation of spontaneous lung SCC associated with increased inflammation (Xiao et al., 2013). IKKα reduction has been reported in human skin, lung, oral, esophageal, nasopharyngeal, and head and neck SCCs (Liu et al., 2006; Maeda et al., 2007; Marinari et al., 2008; Xia et al., 2013; Yan et al., 2014). HESCCs frequently acquire increased EGFR activity and decreased p53, p16, and Rb expression (Lin et al., 2015; Stoner and Gupta, 2001). However, the role of IKKα in ESCC has not been explored.
In this study, we report that kinase-dead Ikkα knock-in (IkkαKA/KA) mice develop impaired central tolerance, autoinflammation, chronic fungal infection, and ESCC. We demonstrate that autoreactive CD4 T cells permit fungal infection and that inflammation and EGFR activity facilitate chronic fungal infection at the mucosal surface of the epithelium. Fungal infection indeed promotes mouse ESCC (MESCC) development. HESCC from non-APECED patients and MESCC share many hallmarks. Therefore, these findings indicate that chronic fungal infection is an HESCC promoter and highlight avenues for HESCC prevention and treatment.
RESULTS
IkkαKA/KA Mice Develop ESCC Associated with Acquired Chronic Fungal Infection
We observed that C57BL/6 kinase-dead IkkαKA/KA (Zhu et al., 2007) developed esophageal epithelial hyperplasia, and approximately 20% of IkkαKA/KA mice at 5 months of age developed ESCC (Figure 1A). With increasing age, the morbidity of ESCC in IkkαKA/KA mice was increased. After 5 months, some IkkαKA/KA mice started to die due to developing systemic inflammation. These MESCCs expressed HESCC traits (Song et al., 2014; Stoner and Gupta, 2001), including a basal cell marker K5, significantly increased EGFR and STAT3 activities, elevated ΔNp63 expression, and downregulated tumor suppressor p16, p53, and IKKα expression (Figures 1A, 1B, and S1A). The K44A mutation destabilizes the IKKα protein in IkkαKA/KA mice (Figure 1B) (Xiao et al., 2013). IKKα regulates keratinocyte differentiation and proliferation independent of its kinase activity (Cao et al., 2001; Hu et al., 2001; Liu et al., 2008), indicating that a reduced amount of kinase-dead IKKα should not have a dominant negative activity in the esophageal tumorigenesis.
Figure 1. Esophageal SCCs and Fungal Infection in IkkαKA/KA Mice.
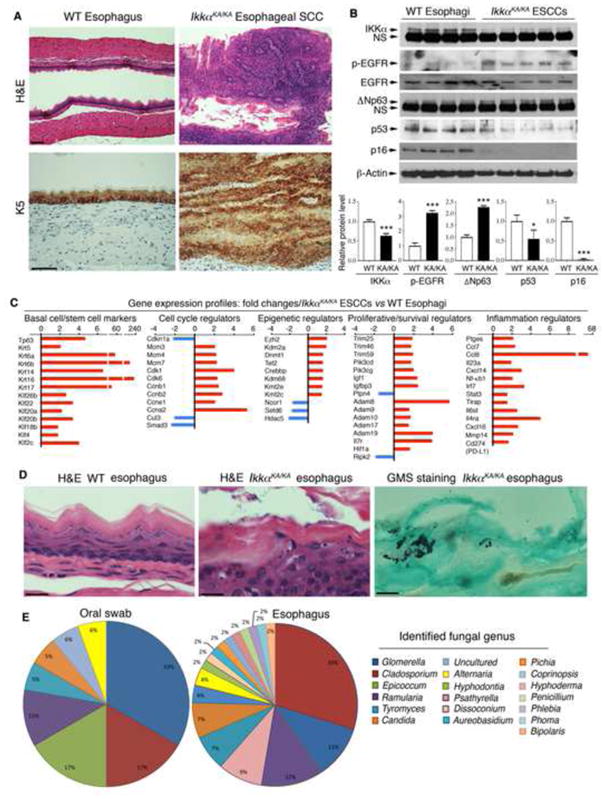
(A) H&E– and immunohistochemistry (IHC)–K5-stained esophagi of 5-month-old WT and IkkαKA/KA mice. K5, keratin 5. Scale bar, 50 μm.
(B) Immunoblotting (IB) shows (top) indicated protein levels in 4 WT esophagi and 5 IkkαKA/KA esophageal SCCs (ESCCs). β-Actin, protein-loading control. Intensities of IKKα, ΔNp63, p53, and p16 normalized by β-Actin and p-EGFR levels normalized by EGFR. Data represent means±SEM (3 repeats, bottom). *, p < 0.05; ***, p < 0.001, t test.
(C) Gene-expression profiles (fold changes): IkkαKA/KA ESCCs versus (vs) WT esophagi using microarray (See Figure S1B and S1C).
(D) H&E–stained esophagi of WT and IkkαKA/KA mice. Scale bar, 20 μm. Right panel: Grocott’s methenamine silver (GMS)–stained fungi. Positive, black spots. Scale bar, 5 μm.
(E) Fungal genera identified by sequencing from cultured colonies (see Figure S1E).
See also Figure S1 and Tables S1 and S2.
To determine a broad spectrum of molecular alterations that may drive the formation of MESCC, using an Array analysis, we found many molecular changes in MESCCs compared to wild-type (WT) esophagi (NCBI: GSE80005), which were also reported in HESCCs (Song et al., 2014) www.cbiportal.org) (Figure S1B and Table S1). These MESCCs highly expressed SCC hallmarks p63, K5/K14, and K6/K16, and expressed significantly increased or decreased levels of genes encoding proteins that regulate stem cell properties, cell cycle, DNA replication, epigenetic mechanisms, cell proliferation or survival, oxidative stress, DNA repair, inflammatory pathways, and PD-L1 (Figures 1C, S1C, and Table S2). Among these molecular alterations, EGFR, STAT3, Adams, ΔNp63, Smad3, and Trims are IKKα targets (Descargues et al., 2008; Liu et al., 2011; Liu et al., 2008).
In hematoxylin and eosin (H&E)–stained sections, we noticed fungus-like microorganisms on the surface of the esophagus in IkkαKA/KA mice, but not in WT mice (Figure 1D). The microorganisms were stained with Grocott’s methenamine silver (GMS), a stain used for fungi (Figure 1D). Oral swabs and ground esophagus extracts of IkkαKA/KA mice, but not WT mice, generated a high yield of fungal colonies in yeast peptone dextrose (YPD) agar plates containing the antibiotic chloramphenicol (Figure S1D). Sequencing of amplicons with ITS1-ITS4 and ITS1-ITS2 fungal-specific PCR primers allowed us to identify 19 different species of fungi from these colonies. These fungi belong to Ascomycota and Basidiomycota phyla (Figures 1E and S1E). C. cladosporioides and G. lagenaria were two major fungal species that colonized in the oral cavities and esophagi of IkkαKA/KA mice, suggesting that the fungi may have spread from the oral cavity to the esophagus. Using a direct sequencing approach, we detected an additional fungus, Fusarium sp. in IkkαKA/KA mice, but did not detect C. albican that was reported in human HESCC. Likely, it is because our mice and humans live in different environments. Cladosporium, Epicoccum, Candida (tropicalis), Alternaria, Aureobasidium, Penicillium, Fusarium, and Phoma genera were reported in human oral cavities or human cancers (Ghannoum et al., 2010; Underhill and Iliev, 2014). C. cladosporioides is one of the most common fungi both indoors and outdoors and can be pathogenic (Polack et al., 1976). In addition, we detected a low number of fungal colonies in the small intestine and colon of some IkkαKA/KA mice but none in WT mice (Figure S1F). We did not detect fungal colonies from the lungs and skin. Because we observed no tumors in the colon, we further investigated the activity of chronic fungal infection in esophageal tumorigenesis.
IkkαKA/+ and WT mice did not develop any phenotypes or chronic fungal infection after co-housing with IkkαKA/KA mice for more than 5 months. IkkαKA/KA female mice were unable to breed. Thus, IkkαKA/+ females were used to produce WT, IkkαKA/+, and IkkαKA/KA mice. We started to detect increased fungal infection in the oral cavities of IkkαKA/KA mice at 6–7 weeks of age. Likely, IkkαKA/KA mice acquired fungal infection from the environment.
IkkαKA/KA Mice Develop Impaired Central Tolerance and Systemic Inflammation
A marked infiltration of inflammatory cells was present in the pancreases, lungs, intestines, livers, kidneys, salivary glands, eyes, thyroids, and multiple stratified epithelial organs in IkkαKA/KA mice older than four to five months, compared to WT mice (Figures S2A and S2B, and data not shown). By using immunofluorescence (IF) staining, the serum from IkkαKA/KA mice, but not from WT mice, reacted with the kidneys, salivary glands, and pancreases of Rag1−/− mice (Figure S2C), indicating that IkkαKA/KA mice developed systemic inflammation and autoantibodies.
We then hypothesized that the autoimmune phenotype of IkkαKA/KA mice may be secondary to a defect in central tolerance development (Lomada et al., 2007). We found a markedly decreased extension of the lightly stained medullary region that was filled with an increased number of small lymphocytes in H&E–stained sections of IkkαKA/KA thymuses, compared to WT (Figure 2A). IF staining verified that, compared to WT, IkkαKA/KA medullary region had a reduced number of mTECs as identified by K5, Aire, and lectin Ulex europaeus agglutinin-1 (UEA-1)–binding cells (Figure 2A). The intensity of Aire staining was reduced in IkkαKA/KA cells compared to WT. A frequency of CD11c+ dendritic cells was decreased in the IkkαKA/KA thymus compared to the WT (Figure S2D). The number of CD4+CD25+Foxp3+ regulatory T cells (Tregs) was reduced in IkkαKA/KA thymuses and spleens compared to WT (Figure 2B), which was presumably the result of a defect in negative T-cell selection. Thus, central tolerance development is impaired in IkkαKA/KA mice.
Figure 2. mTEC Is Essential for Preventing Chronic Fungal Infection and Esophageal Phenotypes.
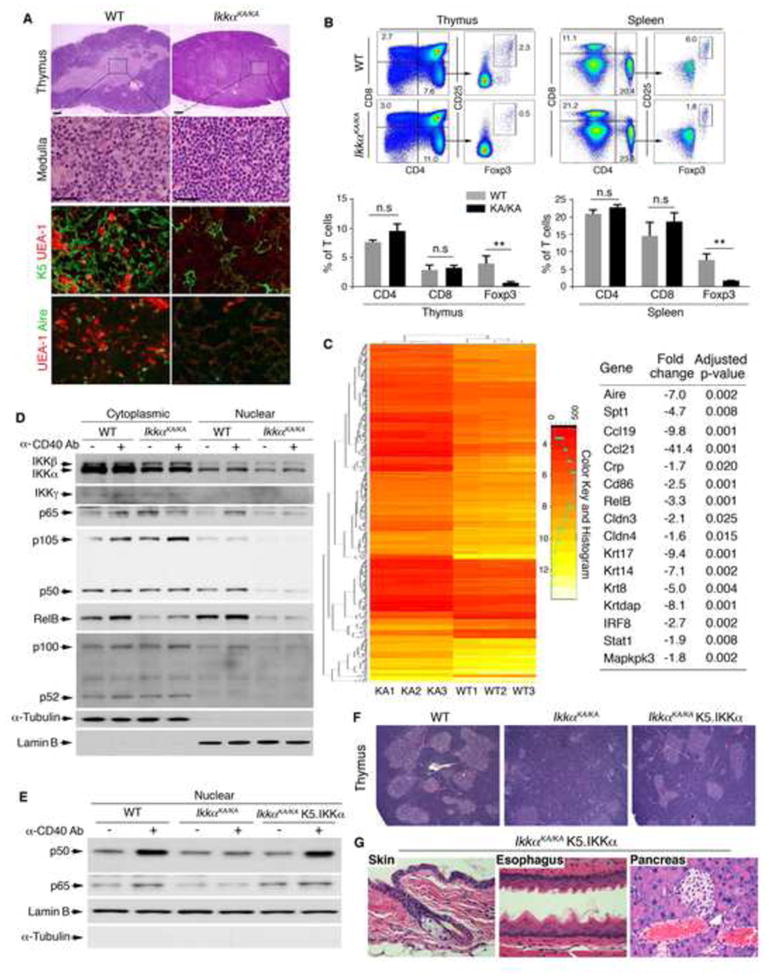
(A) H&E–stained thymuses of WT and IkkαKA/KA mice at 6 weeks of age. Lighter-stained areas are medullary regions. Boxes and lines in row one indicate amplified, lighter-stained medullary regions. Immunofluorescence (IF) staining for K5/UEA-1 in row three and UEA-1/Aire mTECs in row four. Scale bar, 50 μm.
(B) Flow cytometry analyzes CD4, CD8, and CD4+CD25+Foxp3+ Treg cells from thymuses and spleens of IkkαKA/KA and WT mice at 6 weeks of age (top). Data represent means±SEM (3 repeats) (bottom). **, p < 0.01; n.s, not significant, t test; KA/KA, IkkαKA/KA.
(C) Gene-expression profiles of mTEC mRNA of IkkαKA/KA (KA, n =3) and WT mice (n = 3). The heat map contains 600 genes. Spt1, salivary protein 1; Crp, C-reactive protein; Cldn, claudin; Krt, keratin.
(D) IB shows indicated cytoplasmic and nuclear protein levels in mTECs of 4 WT and 8 IkkαKA/KA mice following anti-CD40 antibody treatment. α-Tubulin, cytoplasmic protein-loading control; Lamin B, nuclear protein-loading control.
(E) IB shows nuclear p50 and p65 levels in mTECs from 4 WT, 8 IkkαKA/KA, and 4 IkkαKA/KA;K5.IKKα mice following anti-CD40 antibody treatment. Lamin B, nuclear protein-loading control. Loading controls see (D).
(F) H&E–stained thymuses of WT, IkkαKA/KA, and IkkαKA/KA;K5.IKKα mice. Scale bar, 40 μm.
(G) H&E–stained organs of IkkαKA/KA;K5.IKKα mice. Scale bar, 40 μm.
See also Figure S2.
mTEC Development is Essential to Preventing Fungal Infection
To determine the molecular mechanism behind IKKα regulation of mTEC development, we analyzed gene-expression profiles. We found a downregulation in expression of genes encoding regulators for central tolerance development (Hamazaki et al., 2007; Lomada et al., 2007), including Aire; Aire’s target, TRAs: salivary protein 1, C-reactive protein, and fatty acid–binding protein; mTEC-expressed molecules: CD86, claudin3, and claudin4; several keratins; and molecules involved in cell survival and mitogenic pathways in IkkαKA/KA mTECs compared to WT (Figure 2C, NCBI: GSE52137). Using RT-PCR, we verified the expression of these genes in IkkαKA/KA and WT mTECs (Figure S2E). These molecular alterations may contribute to the defect in mTEC development in IkkαKA/KA mice.
Both canonical and noncanonical NF-κB activities are required for mTEC development and Aire expression (Akiyama et al., 2008). Whether IKKα regulates the canonical NF-κB pathway in mTECs was unknown. Treatment with an anti-CD40 antibody (an agonist) increased nuclear p65/p50 levels in the mTECs of WT mice, but not in IkkαKA/KA mice (Figure 2D), indicating a defect in the CD40-induced canonical NF-κB pathway in IkkαKA/KA mTECs. IKKα expression was reduced in IkkαKA/KA mTECs compared to WT. Expression of IKKβ and IKKγ in WT and IkkαKA/KA cells was similar. Treatment of mice with an anti-LTβR antibody (an agonist) induced an increase in nuclear p52 levels but did not alter p100 levels in WT cells. In contrast, in IkkαKA/KA cells, p52 and RelB levels were markedly decreased, and the agonist was not able to induce p52 nuclear translocation, causing increased p100 levels (Figures S2F and S2G). These results suggest that kinase-dead IKKα impairs nuclear translocation of both p50:p65 and p52:RelB NF-κB, resulting in a defect in mTEC development.
Keratin 5 (K5) is specifically expressed in mTECs. To verify the effect of epithelial-specific IKKα on NF-κB activities, mTEC development, and fungal infection, we introduced the K5.IKKα transgene, in which IKKα cDNA was overexpressed under the control of an epithelial cell–specific K5 promoter (Liu et al., 2008), into IkkαKA/KA mice. Unlike IkkαKA/KA cells, mTECs from IkkαKA/KA;K5.IKKα transgenic mice showed an increase in CD40-induced nuclear p50 and p65 translocation (Figure 2E). Also, K5.IKKα partially restored the size of the medullary regions and Treg numbers in the thymus, abolished the systemic inflammation and malignant phenotypes, and eliminated fungal infection in IkkαKA/KA;K5.IKKα mice (Figures 2F, 2G, S2H, and S2I). IkkαKA/KA;K5.IKKα mice lived for more than a year, maintaining an almost-normal phenotype. These results demonstrate that IKKα plays an essential role for activation of NF-κB pathways required for mTEC development. IKKα is also expressed in the basal squamous epithelial cells and its loss may contribute to the fungal growth on the mucosal surface of the esophageal epithelium and epithelial phenotypes.
TCR Vβ5.1 Repertoire Cells Are Increased in IkkαKA/KA Mice and K5.IKKα Suppresses the T-Cell Repertoire
Flow cytometric analysis and IF staining verified that K5.IKKα restored numbers of K5+UEA-1+ and CD45+integrinα6+UEA-1+ mTECs in IkkαKA/KA;K5.IKKα thymuses compared to IkkαKA/KA thymuses (Figures 3A and S3A). Expression of LTβR and CD80 was decreased in IkkαKA/KA mTECs compared to WT and K5.IKKα reversed this defect (Figure S3B), suggesting that K5.IKKα regulates the expression of the genes that are important for mTEC development. As mTECs determine the thymic microenvironment for selecting TCR repertoires (Akiyama et al., 2005), we examined the TCR Vβ repertoire including Vβ2, 3, 4, 5.1/5.2, 6, 7, 8.1/8.2, 8.3, 9, 10b, 11, 12, 13, 14, and 17a. The CD4+ and CD8+ T-cell populations expressing increased Vβ5 were detected in the IkkαKA/KA thymus and spleen compared to WT (Figures 3B–3D). Reintroduced K5.IKKα reduced Vβ5-positive T cells. We did not detect significant alterations in other repertoires. Because the Vβ5 antibody recognizes Vβ5.1 and Vβ5.2, we used PCR to verify increased Vβ5.1 and Vβ5.2 expression in T cells of IkkαKA/KA mice compared to WT and that K5.IKKα decreased Vβ5.1 and Vβ5.2 expression (Figures 3E and S3C). Increased Vβ5.1 repertoire was detected in the T cells from two sibling patients with APECED who carried AIRE mutations and fungal infection (Kogawa et al., 2002), suggesting that IkkαKA/KA T cells carry a signature of increased Vβ5.1 repertoire shared with APECED patients.
Figure 3. IKKα Regulates mTEC and TCR Repertoire; T Cells are Autoreactive in IkkαKA/KA Esophagi.
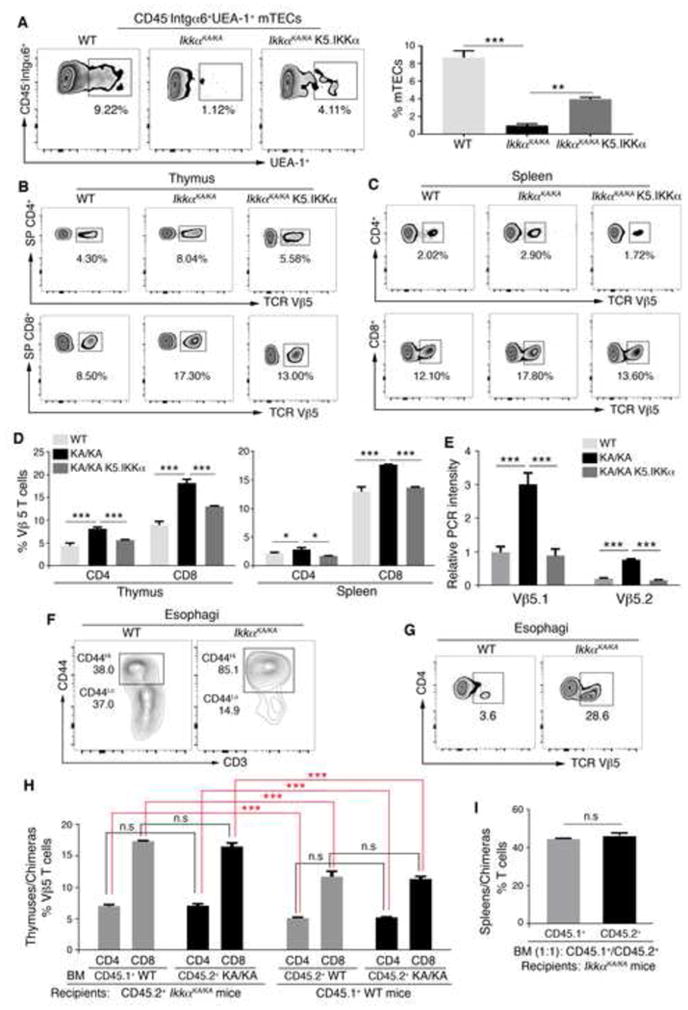
(A) Flow cytometry analyzes mTECs gated at CD45−Integrinα6+(Intgα6)UEA-1+ cells from thymuses of WT, IkkαKA/KA, and IkkαKA/KA;K5.IKKα mice at 8 to 10 weeks of age (left). Data represent mean±SEM (3 repeats, right). **, p < 0.01; ***, p < 0.001, one-way ANOVA.
(B–D) Flow cytometry analyzes TCR Vβ5 repertoire in T cells of the thymus and spleen of WT, IkkαKA/KA, and IkkαKA/KA;K5.IKKα mice at 8 to 10 weeks of age (B–C). SP, single positive. Data represent mean±SEM (D, 3 repeats). *, p < 0.05; ***, p < 0.001, one-way ANOVA.
(E) PCR for expression of Vβ5.1 and Vβ5.2 TCR repertoire in splenic T cells of mice. Gapdh is used to normalize Vβ5.1 and Vβ5.2 levels. Data represent mean±SEM (3 repeats). ***, p < 0.001, one-way ANOVA.
(F–G) Flow cytometry analyzes CD3+CD44Hi and CD3+CD44Lo cells (F), and TCR Vβ5 positive CD4+ T cells (G) gated at CD45+ cells in esophagi of 8 WT and 4 IkkαKA/KA mice at 5 months of age.
(H) Flow cytometry analyzes TCR Vβ5 repertoire positive cells gated at CD4 or CD8 from the thymus of chimeric mice. WT or IkkαKA/KA (KA/KA) BM was injected to KA/KA mice (left). WT BM or IkkαKA/KA (KA/KA) BM was injected to WT mice (right). Data represent mean±SEM (3 repeats). ***, p < 0.001; n.s, not significant, t test.
(I) Flow cytometry analyzes CD3 T cells from spleens of chimeric mice. Data represent mean±SEM (3 repeats). n.s, not significant, t test.
See also Figure S3.
Most T cells found in IkkαKA/KA esophagi were CD4 cells (Figure S3D) and 85% of these T cells expressed activated- or memory-cell makers (CD3+CD44Hi) in IkkαKA/KA esophagi compared to WT (Figure 3F). Notably, the TCR Vβ5 cell population was amplified in IkkαKA/KA esophagi compared to IkkαKA/KA thymuses and spleens (Figures 3B, 3C, 3G, S3E, and S3F). These results indicate that T cells in IkkαKA/KA esophagi are autoreactive.
To confirm whether the thymic microenvironment regulates T-cell development, we performed bone marrow (BM) transplantation using C57BL/6 IkkαKA/KA mice expressing a CD45.2+ marker and congenic C57BL/6 WT mice expressing a CD45.1+ marker. Irradiated IkkαKA/KA mice receiving IkkαKA/KA BM and irradiated WT mice receiving WT BM were used as controls. At 5 weeks after BM transplantation, the Vβ5 T-cell population was similar in the thymuses of chimeric IkkαKA/KA mice receiving WT BM or IkkαKA/KA BM and chimeric WT mice receiving WT BM or IkkαKA/KA BM (Figures 3H and S3G), indicating that the thymic microenvironment regulates TCR repertoire development. To examine whether this effect is induced by a T cell–intrinsic activity or not, we performed another transplantation and found that after irradiated IkkαKA/KA mice received mixed IkkαKA/KA and WT BM cells at a 1:1 ratio, the ratio of CD45.2+ and CD45.1+ cells from chimeric IkkαKA/KA mice remained similar (Figure 3I), demonstrating that IkkαKA/KA thymic microenvironment impairs T-cell development.
CD4 T-Cell Deletion or Antifungal Drug Treatment Prevents Fungal Infection and ESCC in IkkαKA/KA Mice
Impaired central tolerance mediates self-reactive T cell–associated autoimmunity. Next, we attempted to understand the role of autoreactive T cells in fungal infection, inflammation, and esophageal tumorigenesis. The numbers of infiltrating macrophages (F4/80) and T cells (CD3+) were markedly increased in IkkαKA/KA esophagi compared to WT (Figures 4A and S4A, top panel). All 20 IkkαKA/KA;Rag1−/− mice that lack both T and B cells did not show esophageal epithelial phenotypes, oral fungal infection, systemic inflammation, and other organ phenotypes (Figures 4A and S4B, and data not shown). Similar results were obtained in all 20 IkkαKA/KA;Cd4−/− mice, although these mice did show a few infiltrating inflammatory cells in some organs (Figures 4A, S4B, and S4C). The results suggest that IkkαKA/KA autoreactive T cells are associated with increased fungal infection, macrophage recruitment, and esophageal carcinogenesis. Then, we treated mice at 8 weeks of age with amphotericin B, an antifungal drug, for 3 months and monitored fungal infection by culturing oral swabs. The treatment significantly reduced the fungal burden, prevented macrophage infiltration, rescued esophageal phenotypes in 70% of IkkαKA/KA mice, and abolished esophageal tumors (Figures 4A–4C). The antifungal treatment resolved fungal infection better in the mouth than in the esophagus. The antifungal therapy did not rescue the mTEC defect in IkkαKA/KA mice (Figure 4A), suggesting that decreased fungal burden reduces inflammation and esophageal carcinogenesis.
Figure 4. T-Cell Depletion or Antifungal Drug Treatment Diminishes Inflammation and Malignancies in IkkαKA/KA Esophagi.
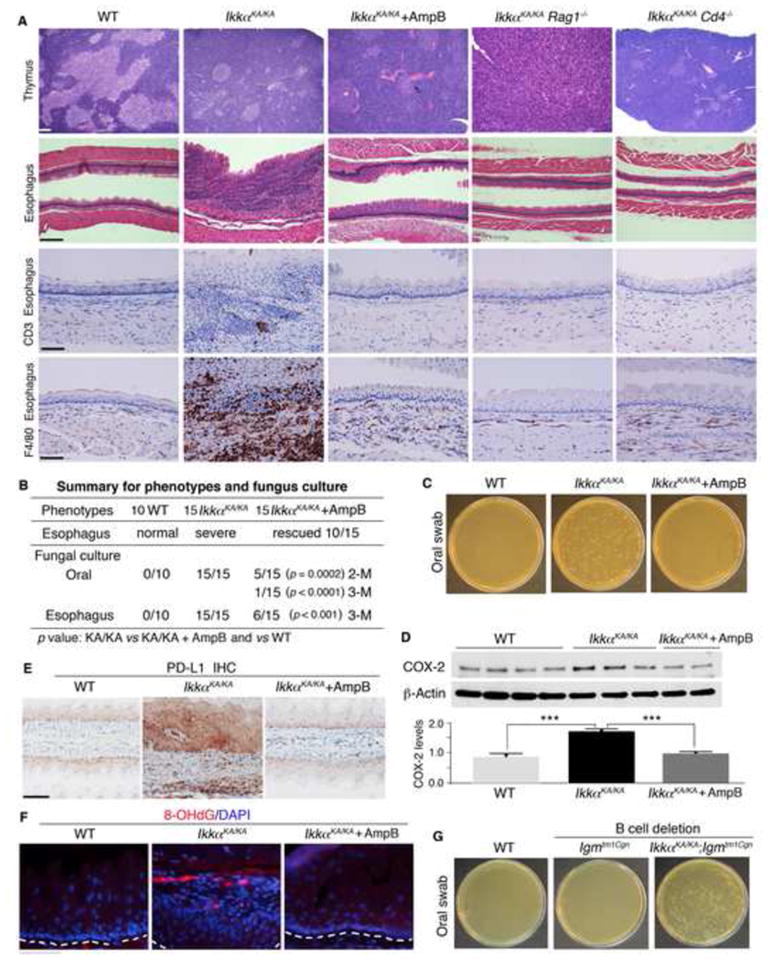
(A) H&E–stained thymuses and esophagi of WT, IkkαKA/KA, antifungal-treated IkkαKA/KA, IkkαKA/KA;Rag1−/−, and IkkαKA/KA;Cd4−/− mice at 5 months of age (Scale bar, 50 μm) and IHC–stained esophagi with the anti-CD3 or anti-F4/80 antibody. AmpB, antifungal drug: amphotericin B; positive, brown color; IkkαKA/KA;Rag1−/− and IkkαKA/KA;Cd4−/− mice (n = 20/group). Scale bar, 30 μm.
(B) A summary of esophagus phenotypes (severe inflammation and malignancy) from indicated mice. Fungus detection in oral swabs and esophagus extracts of indicated mice was shown; number, mouse numbers; M, treatment months. Fisher’s exact test was applied for statistical analyses (p value).
(C) Fungal cultures from oral swabs of WT, IkkαKA/KA, and antifungal-treated IkkαKA/KA mice (see 4B). Data is representative of YPD agar plates containing chloramphenicol (10 μg/ml).
(D) IB shows COX-2 levels in esophagi of 4 WT, 3 IkkαKA/KA, and 2 AmpB-treated IkkαKA/KA mice and their intensities analyzed by one-way ANOVA. ***, p < 0.001. β-Actin, protein-loading control.
(E) PD-L1–IHC-stained esophagi of WT, IkkαKA/KA, and AmpB-treated IkkαKA/KA mice (n = 5/group). Scale bar, 30 μm.
(F) IF staining detected 8-OHdG in esophageal squamous epithelial cells of WT, IkkαKA/KA, and antifungal-treated IkkαKA/KA mice (n = 3/group). White lines are located beneath esophageal basal cell layers. DAPI, the nucleus of cells. Scale bar, 30 μm.
(G) Fungal cultures from the oral swabs of WT, Ighmtm1Cgn (B-cell–lacking), and IkkαKA/KA;Ighmtm1Cgn mice at 10 to 12 weeks of age. Data is representative of YPD agar plates (n = 5/group).
See also Figure S4.
Several studies reported increased cyclooxygenase-2 (COX-2) and PD-L1 expression in the vast majority of HESCCs (Chen et al., 2014; Zimmermann et al., 1999). Consistently, increases in COX-2 and PD-L1 expression as well as increased PD-L1–positive macrophages were detected in IkkαKA/KA esophagi compared to WT, and antifungal treatment reduced COX-2 and PD-L1 expression (Figures 4D, 4E, and S4A, bottom panel). We observed that the malignant epithelia expressing increased PD-L1 expression were tightly associated with infiltrating cells (Figure 4E). We did not see a marked increase in PD-L1 expression in other epithelial organs of IkkαKA/KA mice, suggesting that elevated PD-L1 expression in IkkαKA/KA esophagi is likely induced by the esophageal microenvironment. We detected an increased DNA damage marker, 8-OHdG, in IkkαKA/KA esophageal epithelia, and antifungal treatment reduced DNA damage (Figure 4F), suggesting that increased fungal burden increases DNA damage.
The absence of B cells in IkkαKA/KA;Igmtm1Cgn mice did not rescue the inflammatory and malignant phenotypes, nor did it reduce fungal burden (Figures 4G, S4D, and S4E), suggesting that B cells are not a major cause for increased fungal burden and inflammation in the esophagi of IkkαKA/KA mice.
WT T Cells Eliminate Fungal Infection, Inflammation, and Esophageal Malignancy
To determine the effect of T cells on fungal infection, inflammation, and ESCC, we transferred WT and IkkαKA/KA T cells (5 × 106 cells per injection for two injections) into 6 to 7-week-old IkkαKA/KA;Rag1−/− mice. Lymphopenic mice do not develop increased fungal infections (Ostanin et al., 2007). Transferring IkkαKA/KA T cells but not WT T cells induced marked macrophage infiltration and fungal colonization on the esophagi of all IkkαKA/KA;Rag1−/− mice (not in Rag1−/− mice) at 4 months after the transfer. Two of 5 recipient IkkαKA/KA;Rag1−/− mice developed esophageal malignancies (Figures 5A and S5A). Repeated experiments verified that the incidence of the esophageal malignancy in IkkαKA/KA;Rag1−/− mice receiving IkkαKA/KA T cells was significantly higher than IkkαKA/KA;Rag1−/− mice receiving WT T cells (p = 0.017, n = 10 mice/each group, Fisher’s exact test). Transferring IkkaKA/KA or WT B cells (5 × 106 cells per injection) did not induce severe phenotypes in 10 of 10 IkkαKA/KA;Rag1−/− mice (Figure S5B). These results indicate that autoreactive IkkαKA/KA T cells permit fungal infection, resulting in inflammation and ESCC.
Figure 5. IkkαKA/KA Autoreactive T Cells and Fungal Infection Contribute to Esophageal Carcinogenesis.
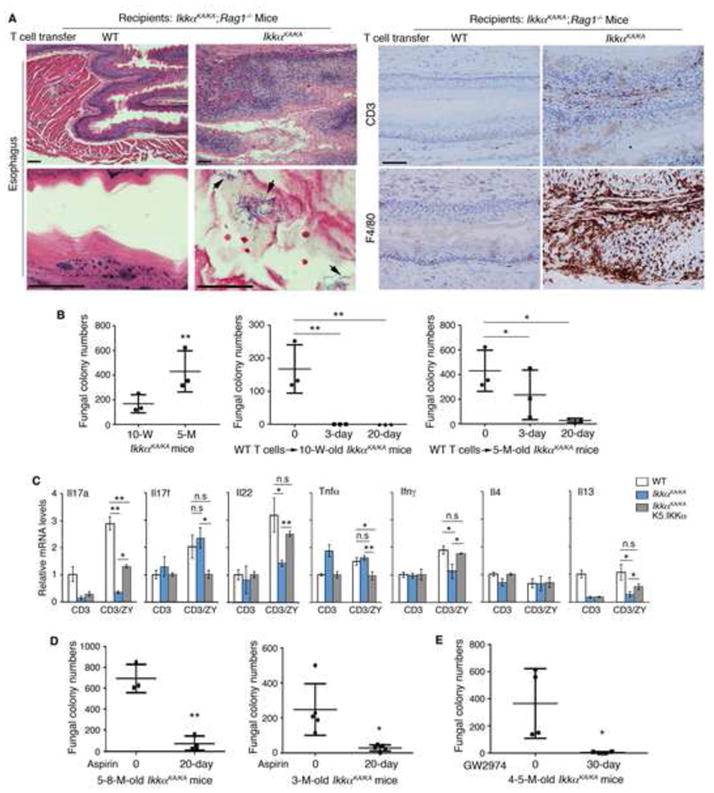
(A) Left: H&E–stained esophagi of IkkαKA/KA;Rag1−/− mice receiving WT or IkkαKA/KA T-cell transfer (n = 5/group, 2 repeats). Arrows indicate fungi. Right: IHC-stained esophagi of IkkαKA/KA;Rag1−/− mice receiving WT or IkkαKA/KA T cells with anti-CD3 and F4/80 antibodies. Scale bar, 30 μm.
(B) Fungal colony numbers in YPD agar plates from oral swabs of IkkαKA/KA mice and IkkαKA/KA mice receiving WT T-cell injection at day 3 and day 20. M, months of age. Data represent mean±SEM (n = 3/group). *, p < 0.05; **, p < 0.01, one-way ANOVA.
(C) Relative mRNA levels of cytokines in T cells from the spleens of WT, IkkαKA/KA, and IkkαKA/KA;K5.IKKα mice following treatment with an anti-CD3 antibody (CD3) only or anti-CD3 antibody and zymosan A (ZY, 2 μg/ml). Data represent mean±SEM (n = 3/group). *, p < 0.05; **, p < 0.01; n.s, not significant, t test.
(D) Fungal colony numbers in YPD agar plates from oral swabs of IkkαKA/KA mice and IkkαKA/KA mice treated with aspirin for 20 days. M, months of age. Data are analyzed by t test (n = 3 for 5- to 8-month-old mouse group; n = 5 for 3-month-old mouse group). *, p < 0.05; **, p < 0.01.
(E) Fungal colony numbers in YPD agar plates from oral swabs of IkkαKA/KA mice and IkkαKA/KA mice treated with GW2974 (0.6 mg/mouse/day), 5 times per week for 30 days. M, months of age. Data are analyzed by t-test (n = 4/group). *, p < 0.05.
See also Figure S5.
To test a therapeutic benefit of WT T cells, we injected WT or IkkαKA/KA T cells (5 × 106 cells per injection) into 5-month-old and 10-week-old IkkαKA/KA mice twice during a two-week interval. At day 20 after the first injection, we found that WT T cells, but not IkkαKA/KA T cells, markedly reduced fungal colony numbers from oral swabs (Figure 5B). This inhibition of WT T cells was more efficient for young IkkαKA/KA mice than for old mice (Figure 5B), likely because the old mice had already developed a more severe fungal infection compared to the young mice. Histological examination consistently showed reduced numbers of infiltrating F4/80 and CD3 cells in the esophagi after WT T-cell therapy (Figure S5C and data not shown). These results suggest that WT T cells eliminate fungal burden and inflammation.
T cells produce IL17, IFNγ, and IL13, which can fight against fungi (Coste et al., 2008; Puel et al., 2011; van de Veerdonk et al., 2011). However, expression of these cytokines was lower in IkkαKA/KA T cells than in WT cells after treatment with an anti-CD3 antibody and zymosan A, a fungal extract (Figure 5C). The defect may contribute to initiating fungal colonization in IkkαKA/KA mice. Furthermore, Ifnγ, Il22, Il17a, and Il13 expression was partially rescued in the T cells of IkkαKA/KA;K5.IKKα mice compared to WT and IkkαKA/KA (Figure 5C), suggesting that epithelial-K5.IKKα influences the expression of these genes in IkkαKA/KA T cells. IKKα upregulates Il17a expression in T cells (Li et al., 2011). Thus, K5.IKKα may be unable to fully restore Il17a expression in IkkαKA/KA T cells.
Inflammation or EGFR Activity Increases Fungal Burden
We detected the increased expression of multiple cytokines and chemokines in IkkαKA/KA macrophages compared to WT (Figure S5D). Consistently, IkkαKA/KA esophagi expressed increased cytokines and chemokines compared to WT (Figure S5E). Antifungal treatment decreased inflammation in the esophagi of IkkaKA/KA mice (Figures S5E, S1B, and S1C). We noticed an increase in Il17a expression in IkkαKAKA esophagi compared to WT but no increases in Il17a expression in IkkαKAKA T cells and macrophages (Figures 5C and S5D), suggesting that the esophageal epithelia may account for the increased Il17a expression. Thus, IkkaKA/KA esophagi are inflamed with elevated cytokine/chemokine expression and macrophage numbers. Like APECED patients (Beerli et al., 2014), IkkαKA/KA mice developed increased anti-IL17F autoantibody (Figure S5F).
To examine the effect of inflammation on chronic fungal infection, we treated IkkαKA/KA mice at 3 to 8 months of ages with aspirin, a nonspecific anti-inflammation drug, for 20 days (Figure 5D). Aspirin treatment significantly reduced the number of fungal colonies in oral swabs. We used clodronate-loaded liposomes (Xiao et al., 2013) to deplete macrophages in IkkαKA/KA mice and obtained results similar to those from aspirin treatment (data not shown); however, liposome treatment caused the death of 5 treated IkkαKA/KA mice in a group containing 10 mice. These results suggest that inflammation contributes to the maintenance of chronic fungal infection.
It has been reported that fungi can interact with and activate EGFR and that treatment with an EGFR inhibitor significantly reduces the severity of oropharyngeal candidiasis (Zhu et al., 2012). EGFR is an IKKα target (Liu et al., 2008). We then treated IkkαKA/KA mice with GW2974, an EGFR inhibitor, for 30 days. The treatment significantly reduced fungal burden in oral swabs of IkkαKA/KA mice compared to untreated mice (Figure 5E). Thus, EGFR activation increased fungal burden.
Oral Fungal Infection Promotes Esophageal Tumorigenesis and Fungal Infection is Associated with HESCC
To determine whether increased fungal infection enhances tumorigenesis, we orally infected WT, IkkαKA/+, and IkkαKA/KA mice with Cladosporium cladosporioides (2 × 107 fungi per mouse) every two weeks for a total of 4 infections. After infection, all IkkaKA/KA mice, including untreated IkkaKA/KA mice, maintained oral fungal infection. Fungal colonization in IkkaKA/+ mice required more rounds of infections and WT mice were significantly resistant to fungal infection (Figures 6A and 6B). IkkαKA/KA mice started to die after the fourth infection. Histologic examination revealed that fungus-infected IkkαKA/KA mice developed increased numbers of esophageal or oral tumors that were K5 positive (Figures 6B, 6C, and S6A). These mice also developed hyperkeratosis and hyperplasia in the esophagi and oral cavities (Figure S6A and data not shown). IkkαKA/+ mice were normal. Infected IkkαKA/+ and WT mice developed hyperplasia in the esophagi and oral cavities, and the phenotype was more severe in IkkαKA/+ mice than in WT mice. We observed a robust correlation between increased esophageal phenotypes and increased fungal colonization among these mice. Oral tumors were detected in one of 16 infected IkkαKA/+ mice. Therefore, repeated fungal infections significantly promote the esophageal and oral tumorigenesis and IKKα protects the esophageal and oral epithelium from fungal infection and neoplastic transformation in a dose-dependent manner. We then propose a working model for mouse autoimmune disease, fungal infection, and MESCC development (Figure 6D).
Figure 6. Fungal Infection Promotes Pathogenesis of Mouse Oral and Esophageal Cancers and Fungi are Detected in HESCCs.
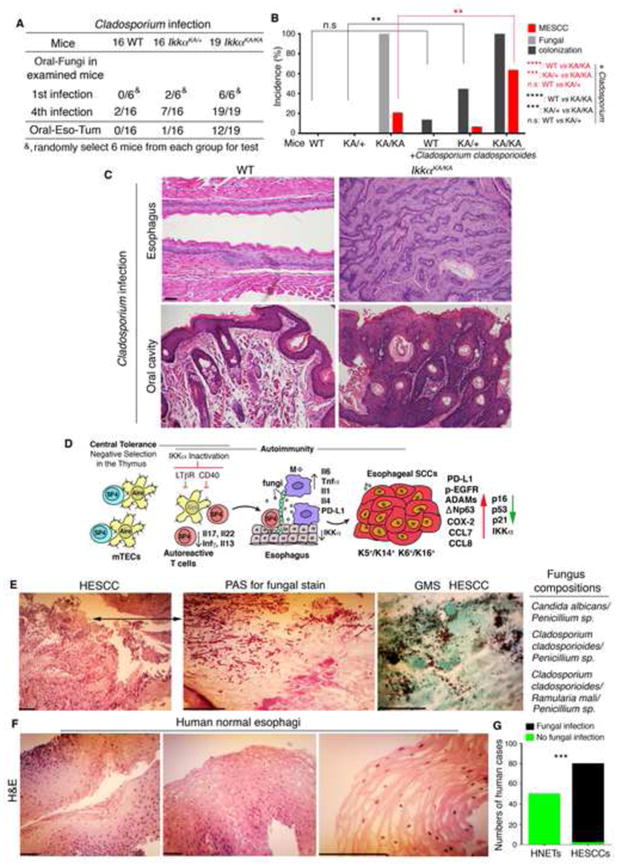
(A–B) Oral fungal infection and tumors detected in WT, IkkαKA/+ (KA/+), and IkkαKA/KA (KA/KA) mice orally infected with fungi (A). Oral-Fungi, monitoring fungi in oral swabs; the first (1st) infection; Oral-Eso-Tum, oral or esophageal tumors; 4th, the fourth infection. Data are analyzed by Fisher’s exact test. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s, not significant (B).
(C) Representative H&E–stained esophagus and oral cavity of WT mice, and esophageal SCC in situ and oral SCC from IkkαKA/KA mice after 4 fungal infections. Scale bar, 50 μm.
(D) A working model of esophageal SCC development promoted by impaired central tolerance mediated autoimmunity, T–cell defects, inflammation, fungal infection, and IKKα reduction. mTECs, medullary thymic epithelial cells; SP4, CD4 single-positive T cell; MΦ, macrophage; lines with circles on mTEC surfaces are tissue-restricted antigens; Aire, Aire expression in the nucleus of mTECs; multiple crossed lines in the nuclei of tumor cells, DNA damage. Down arrow, decrease; up arrow, increase; cross lines, inactivation.
(E) Left panels (photos): Human ESCC sections stained with Periodic Acid-Schiff (PAS) staining or GMS for fungal staining. Left panel 1, Scale bar 40 μm; Left panels 2 and 3, Scale bar 5 μm. An arrow indicates the area that contains PAS-stained fungi. Right panel: Identified fungal species from paraffin sections of HESCC cases by DNA sequencing.
(F) H&E–stained human normal esophageal tissue sections. Scale bar, 40 μm.
(G) Data in Figures 6E and 6F are statistically analyzed by Fisher’s exact test. ***, p < 0.001. HNETs, human normal esophageal tissues = 50 and HESCCs = 80.
See also Figure S6.
HESCC is one of the most common and deadliest malignancies in China and South Asia. We also examined the association between fungal infection and non-APECED-derived HESCCs in a cohort containing 50 human normal esophageal tissue specimens and 80 stage-II and III HESCCs expressing K5 and p63 from Chinese non-APECED patients (Figures S6B, S6C, and S6D). We detected fungi in 78 of 80 (97.5%) human HESCC patients using a light microscope but not in normal human tissues (Figures 6E–6G). The association between fungal infection and HESCCs was statistically significant (Figure 6G), although the severity of the fungal infection was different among these HESCCs. We verified fungi in H&E–stained HESCC slides with Periodic acid-Schiff (PAS) and GMS staining and further confirmed these fungi in the paraffin sections by sequencing (Figure 6E). We extracted DNA from 3 H&E-stained HESCC slides and 2 normal esophagus slides, generated a PCR-product library for each case, and sequenced fungi. We identified 4 species of fungi from 3 HESCC patients, in which each HESCC case contained 2 or 3 different fungi: Candida albicans/Penicillium sp, Cladosporium cladosporioides/Penicillium sp, and Cladosporium cladosporioides/Ramularia mali/Penicillium sp, but not from controls (Figure 6E, right panel). C. albicans has been reported in HESCCs (Rautemaa et al., 2007). Most studies used specific culture media and morphological features to identify fungi in HESCCs (Mermel et al., 2009). The sequencing method we used may be more sensitive than other methods. Both Cladosporium and Penicillium are detected in human mouths (Ghannoum et al., 2010).
To determine shared hallmarks between HESCCs from non-APECED patients and MESCCs derived from IkkαKA/KA mice, we examined a HESCC tissue array containing 60 HESCCs and their adjacent tissues. We found increased numbers of macrophages (CD68) and T cells (CD3) and decreased IKKα levels in the HESCCs compared to the adjacent tissues (Figures 7A–7C and S7A). We often observed reduced IKKα expression in inflamed mouse organs. Thus, we hypothesized that inflammation may downregulate IKKα expression. Western blotting showed reduced IKKα expression in human SCC A431 and NCI 520 cell lines following treatment with TNFα or IFNγ at 72 or at 48 hr and did not show IKKα expression in human SCC-25 cells (Figures 7D and S7B), suggesting that increased cytokines downregulate IKKα expression in human SCC cells although the mechanism remains to be determined. The mutations of CHUK that encodes IKKα were detected in HESCCs (Figure 7E from www.cbiportal.org). Like MESCC, PD-L1 expression was significantly higher in the tumor regions than in the adjacent tissues (Figure 7F). Therefore, both HESCCs and MESCCs bear marked inflammatory cell infiltration, elevated PD-L1 expression, and reduced IKKα levels.
Figure 7. Increased Macrophage and T Cells and PD-L1 Levels, and Reduced IKKα in HESCCs.
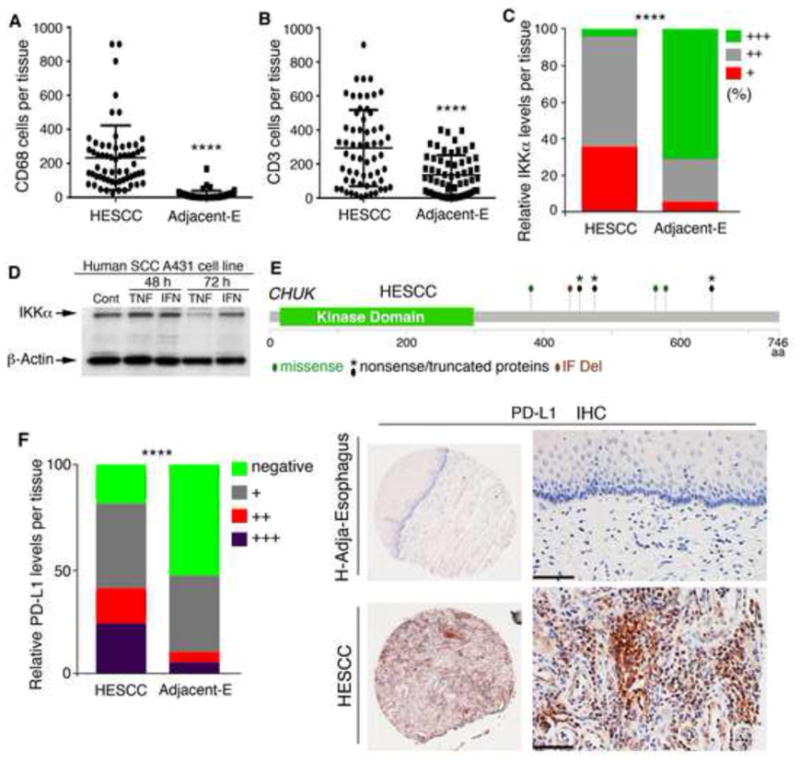
(A) Macrophage (CD68) numbers in the human tissue array containing 60 HESCCs and 60 adjacent tissues (Adjacent-E), as analyzed by t-test. ****, p < 0.0001.
(B) T-cell (CD3) numbers in the human tissue array containing 60 HESCCs and 60 adjacent tissues, as analyzed by t test. ****, p < 0.0001.
(C) Staining intensities of IKKα using IHC with an anti-IKKα antibody in the human tissue array containing 60 HESCCs and 60 adjacent tissues. Staining intensities show SCCs vs adjacent tissues: strong staining (+++), 5% vs 38.3%; medium staining (++), 25% vs. 56.7%; and weak staining (+), 70% vs. 5%. Data are statistically analyzed by Chi-square test for each staining group between SCCs and adjacent tissues. ****, p < 0.0001.
(D) IB shows IKKα levels in human SCCA431 SCC cell line following TNFα (TNF, 10 ng/ml) and IFNγ (IFN, 10 ng/ml) treatment at 48 and 72 hr. Cont, Control.
(E) CHUK mutations in HESCCs obtained from two studies (cbioportal.org). IF, in-frame; Del, deletion.
(F) Left panel: Staining intensities of PD-L1 using IHC with an anti-PD-L1 antibody in the human tissue array containing 60 HESCCs and 60 adjacent tissues. Staining intensities show SCCs vs adjacent tissues. Strong staining (+++) to negative staining (−). Data are analyzed by Chi-square test for each staining group between SCCs and adjacent tissues. ****, p < 0.0001. Right panel: IHC-stained adjacent tissue and HESCC. Scale bar, 40 μm.
See also Figure S7.
DISCUSSION
We demonstrate that defects in the immune system and epithelial cells permit and facilitate fungal infection and that chronic fungal infection promotes MESCC development. A high frequency of fungal infection is detected in HESCCs from non-APECED patients. These MSCCs and HESCCs share many hallmarks. Targeting fungi or epithelial cells that express increased EGFR activity, or correcting immune defects impairs ESCC development. These findings advance our understanding of the relationship between immunity and fungal infection in carcinogenesis and underscore their medical significance for HESCC prevention and treatment.
Chronic Fungal Infection Is an ESCC Promoter
Previously, the consequence of fungal infection in HESCC was not clear. A report (Rautemaa et al., 2007) showed that 6 of 92 APECED patients aged 29–44 years develop oral or esophageal SCCs. These cancer patients at infancy or in the early teenage years prior to SCC formation acquire chronic fungal infection. Like APECED, IkkαKA/KA mice developed chronic fungal infection prior to ESCC. Oral fungal administration promoted whereas antifungal treatment inhibited ESCC development. Therefore, chronic fungal infection promotes esophageal carcinogenesis.
C. albican is frequently reported in HESCCs (Rautemaa et al., 2007), although most studies lack sequence data. We did not detect C. albican in IkkαKA/KA mice. The mice used for this study are maintained in a specific pathogen–free (SPF) facility. These discrepancies are possibly due to different environments where people and mice live or due to a genotype. The effect of C. albican and other fungi on ESCC development needs to be tested.
The microflora of IkkαKA/KA mice did not transfer phenotypes to IkkαKA/+ and WT mice, indicating that a genotype determines this disease. Our unpublished data showed that treatment with antibiotics caused severe rectal prolapse with bleeding in IkkαKA/KA mice. This clinical problem is likely associated with mouse autoimmune conditions. The fecal flora is similar but the oral flora is distinct between IkkαKA/KA and WT mice. The bacterial burden is positively correlated with the fungal burden in the mouths of IkkαKA/KA mice. The detailed relationship between bacteria and fungi in carcinogenesis will be investigated in the future.
Critical Factors That Foster Chronic Fungal Infection
Enormous studies have provided insight into a cause of increased fungal infection by defects in immune system or cytokine products (Puel et al., 2011; Underhill and Iliev, 2014). However, the conditions that recapitulated fungus-associated ESCC development were previously unknown. In this study, we demonstrated that an impaired thymic microenvironment generated autoreactive T cells that permitted chronic fungal infection and incited tissue injury and inflammation. EGFR activation is associated with oropharyngeal candidiasis (Zhu et al., 2012). EGFR, an IKKα target, is an oncogene that can drive SCC development (Liu et al., 2008). Furthermore, we showed that autoreactive T cells alone were not sufficient to induce epithelial phenotypes, for example, IkkαKA/KA T cells induced phenotypes in IkkαKA/KA;Rag1−/− mice but not in Rag1−/− mice. Treatment with aspirin or an EGFR inhibitor reduced fungal burden in IkkαKA/KA mice. Macrophages activate EGFR (Xiao et al., 2013). Therefore, increased EGFR activity and inflammation provide the missing pieces for the establishment of chronic fungal infection in the epithelial organs. The defects in immunity and epithelial cells together facilitate and promote the development of chronic fungal infection and tumorigenesis. APECED is a complicated syndrome with diverse clinical manifestations. Aire deletion alone may not be sufficient to cause defects in immunity and oncogenes/tumor suppressors for generating fungal infection associated-malignant phenotypes in Aire−/− mice. By contrast, IKKα expressed in mTECs and epithelial cells plays a crucial role for preventing fungal infection, autoimmunity, and epithelial phenotypes.
MESCC versus HESCC
APECED is a rare disease; thus, HESCCs from APECED are even more rare (Rautemaa et al., 2007). However, this fact does not reduce the importance of fungal infection in the development of HESCCs from non-autoimmune patients. Most HESCCs are frequently found in China and South Asia (Song et al., 2014), where the weather is moist, humid, and damp, which promotes fungi to thrive. Thus, people who live in these areas may frequently encounter fungi from the environment. Our study revealed a very high frequency of fungal infection in these HESCCs with different degrees of fungal burden. Some patients had a long history of smoking, which induces DNA damage and inflammation. Fungal infection induces Tnfα, Ifnγ, and Il17a expression in the monocytes and T cells of health people and APECED patients although Il17a expression in APECED is impaired (Ryan et al., 2008). We showed that TNFα and IFNγ downregulated IKKα expression. IKKα reduction, was frequently detected in HESCCs. IKKα haplodeficiency promotes carcinogen-induced carcinogenesis (Park et al., 2007). Therefore, fungal infection-induced inflammation and DNA damage can promote IKKα reduction-induced tumorigenesis in the esophagi. Notably, we detected increased T cell and macrophage infiltration and elevated PD-L1 expression in both MESCCs and HESCCs. Although normal T cells fight against fungal infection and tumor cells, PD-L1 blocks the antitumor activity of T cells. How this fungus-involved microenvironment enhances PD-L1 expression remains to be investigated. Clearly, fungus and EGFR can be used for therapeutic targets for HESCC treatment. We are aware of the toxicity of some antifungal drugs after long periods of treatment in humans. Furthermore, IL17A, which can be induced by fungi, plays a role in eliminating fungal infection (Conti et al., 2009). An increased IL17A amount is also pathogenic for human psoriasis (Gordon et al., 2016). Thus, the role of IL17A in ESCCs needs to be tested.
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| IKK alpha antibody, mouse mAb | Novus, clone 14A231 | Cat# NB100-56704 (Alt Cat# IMG-136A) |
| IkB-alpha antibody, mouse mAb | Novus, clone 6A920 | Cat# NB100-56507 (Alt Cat# IMG-127A) |
| Anti-IKKβ antibody, mouse mAb | EMD Millipore, clone 10AG2 | Cat # 05-535 |
| Purified mouse anti-IKKγ, mouse | BD, clone C73-764 (RUO) | Cat# 559675 |
| NFκB p50 antibody (C-19), goat | Santa Cruz | Cat# sc-1190 |
| NFκB p65 antibody (C-20), rabbit | Santa Cruz | Cat# sc-372 |
| IκB-α antibody (C-21), rabbit | Santa Cruz | Cat# sc-371 |
| p53 antibody (FL-393), rabbit | Santa Cruz | Cat# sc-6243 |
| p15/p16 antibody (C-7), mAb | Santa Cruz | Cat# sc-377412 |
| p63 antibody (4A4), mAb | Santa Cruz (discontinued) | Cat# sc-8431 |
| EGFR (1005)-G antibody (1005), rabbit | Santa Cruz | Cat# sc-03 |
| IKKα antibody (M-110), rabbit | Santa Cruz | Cat# sc-7183 |
| RelB antibody (D-4), mouse mAb | Santa Cruz | Cat # sc-48366 |
| LTβR antibody (Ls-14), rat mAb | Santa Cruz | Cat# sc-80167 |
| Lamin B antibody (C-20), goat | Santa Cruz | Cat# sc-6216 |
| NFκB p65 antibody (D14E12), rabbit mAb | Cell Signaling | Cat# 8242 |
| NFκB2 p100/p52 antibody, rabbit | Cell Signaling | Cat# 4882 |
| Phospho-EGF Receptor (Tyr992) antibody, rabbit | Cell Signaling | Cat# 2235 |
| anti-alpha tubulin antibody, rabbit | abcam | Cat# ab4074 |
| Anti-β-actin antibody, mAb | Sigma-Aldrich, clone AC-15, ascites fluid | Cat# A5441 |
| Anti-mouse F4/80 antigen purified, rat mAb | eBioscience, clone BM8 | Cat# 14-4801-81 |
| Anti-Mouse AIRE Alexa Fluor® 488, rat mAb | eBioscience, clone 5H12 | Cat# 53-5934-80 |
| Purified rat anti-mouse CD4, rat | BD, clone RM4-5 (RUO) | Cat# 550280 |
| Anti-CD40 antibody, rat anti-mouse CD40 agonist | From Dr. Giorgio Trinchieri’s lab, clone Fgk115 | N/A |
| Purified NA/LE Hamster Anti-Mouse CD3e | BD, clone 145-2C11 (RUO) | Cat# 553057 |
| Pacific Blue Hamster Anti-Mouse CD3e | BD, clone 500A2 (RUO) | Cat# 558214 |
| PE Rat Anti-Mouse CD4 | BD, clone GK1.5 (RUO) | Cat# 553730 |
| APC-Cy7 Anti-Mouse CD8a | BD, clone 56-6.7 (RUO) | Cat# 557654 |
| FITC Hamster Anti-Mouse CD80 | BD, clone 16-10A1 (RUO) | Cat# 553768 |
| Anti-Mouse CD25 APC | eBioscience, clone PC61.5 | Cat# 17-0251-82 |
| Anti-Mouse/Rat Foxp3 FITC | eBioscience, clone FJK-16s | Cat# 11-5773-82 |
| Anti-Mouse CD3e FITC | eBioscience, clone 145-2C11 | Cat# 11-0031-82 |
| Anti-Human/Mouse CD49f (Integrin alpha 6) PE-Cyanine7 | eBioscience, clone GoH3 | Cat# 25-0495-80 |
| Anti-Mouse CD274 (PD-L1, B7-H1) PE | eBioscience, clone MIH5 | Cat# 12-5982-81 |
| Anti-Mouse Ly-6G (Gr-1) Alexa Fluor 700 | eBioscience, clone RB6-8C5 | Cat# 56-5931-80 |
| Anti-Mouse CD11b PE-Cyanine7 | eBioscience, clone M1/70 | Cat# 25-0112-81 |
| Anti-Mouse CD11c APC-eFluor 780 | eBioscience, clone N418 | Cat# 47-0114-80 |
| Anti-Mouse CD45.1 PE-Cyanine7 | eBioscience, clone A20 | Cat# 25-0453-82 |
| Anti-Mouse CD45.2 eFluor 450 | eBioscience, clone 104 | Cat# 48-0454-82 |
| Bacterial and Virus Strains | ||
| One Shot TOP10 Chemically Competent E.Coli | Invitrogen | Cat# C404010 |
| Biological Samples | ||
| Human esophageal tumor tissue array | US Biomax | Cat# ES1202 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetylsalicylic acid (Aspirin) | Sigma-Aldrich | Cat# A5376 |
| GW2974 | Sigma-Aldrich | Cat# G0668 |
| Zymosan A | Sigma-Aldrich | Cat# Z4250 |
| Phenol-chloroform-isoamyl alcohol | Sigma-Aldrich | Cat# P2069 |
| Chloramphenicol | Sigma-Aldrich | Cat# C0378 |
| YPD Agar | Clontech | Cat# 630410 |
| Amphotericin B for Injection, USP | X-GEN Pharmaceuticals Inc | Cat# NDC 39822-1055-5 |
| TRIzol reagent | Ambion | Cat# 15596026 |
| RT2 SYBR Green qPCR Mastermixes | Qiagen | Cat# 330502 |
| Recombinant Murine IL-17A | Peprotech | Cat# 210-17 |
| Recombinant Murine IL-17F | Peprotech | Cat# 210-17F |
| Recombinant Murine IL-22 | Peprotech | Cat# 210-22 |
| Streptavidin PE | eBioscience | Cat# 12-4517-87 |
| Biotinylated Ulex Europaeus Agglutinin I (UEA I) | Vector Laboratories | Cat# B-1065 |
| Fluorescein labeled Ulex Europaeus Agglutinin I (UEA I) | Vector Laboratories | Cat# FL-1061 |
| Critical Commercial Assays | ||
| GeneChip Mouse Genome 430 2.0 array | Affymetrix | Cat#902491 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat# 78833 |
| Tetro cDNA Synthesis Kit | Bioline | Cat# BIO-65043 |
| RT2 Profiller PCR Array Mouse Cytokines & Chemokines | Qiagen | Cat# PAMM-150ZC |
| EasySep™ Mouse T cell Isolation Kit | STEMCELL | Cat# 19851 |
| Liberase TL | Roche | Cat# 5401119001 |
| Zymoclean™ Gel DNA Recovery Kit | ZYMO Research | Cat# D-4008 |
| YOPO TA Cloning Kit | Invitrogen | Cat# 450641 |
| Mouse Vb TCR screen panel | BD | Cat# 557004 |
| Deposited Data | ||
| Raw data files for microarray | NCBI Gene Expression Omnibus | GSE52137, GSE80005 |
| Experimental Models: Cell Lines | ||
| Human A431 | ATCC | Cat# CRL-1555 |
| Human SCC-25 | ATCC | Cat# CRL-1628 |
| Human NCI-520 | ATCC | Cat# HTB-182 |
| Experimental Models: Organisms/Strains | ||
| Fungus: Cladosporium cladosporioides | This paper | N/A |
| Mouse: IkkaKA/KA, C57BL/6 | Zhu et al., 2007 | N/A |
| Mouse: K5.IKKα, C57BL/6 | Liu et al., 2011 | N/A |
| Mouse: Ikkaflf, C57BL/6 | Liu et al., 2008 | N/A |
| Mouse: K5.Cre, C57BL/6 | Liu et al., 2008 | N/A |
| Mouse: Cd4−/−, C57BL/6, rederived | Jackson Laboratory | Stock# 002663 |
| Mouse: Igmtm1Cgn, C57BL/6, | Jackson Laboratory | Stock# 002288 |
| Mouse: IkkaAA/AA, C57BL/6 | Cao et al., 2001 | N/A |
| Oligonucleotides | ||
| Aire, 5′-GACCTAAACCAGTCCCGGAA-3′ and 5′-ATCCCTTCCACGGCCCCT-3′ | Lomada et al., 2007 | N/A |
| Spt1, 5′-GGCTCTGAAACTCAGGCAGA-3′ and 5′-TGCAAACTCATCCACGTTGT-3′ | Lomada et al., 2007 | N/A |
| Ccl19, 5′-GCTAATGATGCGGAAGACTG-3′ and 5′-ACTCACATCGACTCTCTAGG-3′ | Lomada et al., 2007 | N/A |
| Ccl21, 5′-GCTGCCTTAAGTACAGCCAG-3′ and 5′-GTGTCTGTTCAGTTCTCTTGC-3′ | Lomada et al., 2007 | N/A |
| Fabp, 5′-AGACGGAACGGAGCTCAC-3′ and 5′-GCTCTTCAGCGTTGCTCC-3′ | Lomada et al., 2007 | N/A |
| Gapdh, 5′-GCAGTGGCAAAGTGGAGATT-3′ and 5′-AGAAGGGGCGGAGATGATGA-3 | Zhu, et al., 2007 | N/A |
| Vβ 5.1, forward primer, 5′-AACACTGCCTTCCCTGACCC-3′ | Wettstein, et al., 2008 | N/A |
| Vβ 5.2, forward primer, 5′-GTCTAACACTGTCCTCGCTGATTC-3′ | Wettstein, et al., 2008 | N/A |
| constant region reverse primer, 5′-GCAATCTCTGCTTTTGATGGCT-3′ | Wettstein, et al., 2008 | N/A |
| Real-time qRT-PCR primers:Il6 | Qiagen | Cat# PPM03015A-200 |
| Real-time qRT-PCR primers:Tnfa | Qiagen | Cat# PPM03113G-200 |
| Real-time qRT-PCR primers:Ifng | Qiagen | Cat# PPM03121A-200 |
| Real-time qRT-PCR primers:Il4 | Qiagen | Cat# PPM03013F-200 |
| Real-time qRT-PCR primers:Il13 | Qiagen | Cat# PPM03021B-200 |
| Real-time qRT-PCR primers:Il17a | Qiagen | Cat# PPM03023A-200 |
| Real-time qRT-PCR primers:Il17f | Qiagen | Cat# PPM05398E-200 |
| Real-time qRT-PCR primers:Il22 | Qiagen | Cat# PPM05481A-200 |
| Real-time qRT-PCR primers:Cxcl11 | Qiagen | Cat# PPM03192C-200 |
| Real-time qRT-PCR primers:Gapdh | Qiagen | Cat# PPM02946E-200 |
| Real-time qRT-PCR primers:Kif2c | Qiagen | Cat# PPM04983A-200 |
| Real-time qRT-PCR primers:Cdkn1a | Qiagen | Cat# PPM02901B-200 |
| Real-time qRT-PCR primers:Mcm4 | Qiagen | Cat# PPM03270B-200 |
| Real-time qRT-PCR primers:Mcm7 | Qiagen | Cat# PPM03272E-200 |
| Real-time qRT-PCR primers:Cdk1 | Qiagen | Cat# PPM02907A-200 |
| Real-time qRT-PCR primers:Cdk6 | Qiagen | Cat# PPM02912F-200 |
| Real-time qRT-PCR primers:Ccne1 | Qiagen | Cat# PPM02891B-200 |
| Real-time qRT-PCR primers:Cul3 | Qiagen | Cat# PPM03257F-200 |
| Real-time qRT-PCR primers:Std6 | Qiagen | Cat# PPM26556B-200 |
| Real-time qRT-PCR primers:Dnmt1 | Qiagen | Cat# PPM03685E-200 |
| Real-time qRT-PCR primers:Pik3cg | Qiagen | Cat# PPM03469A-200 |
| Real-time qRT-PCR primers:Igf1 | Qiagen | Cat# PPM03387F-200 |
| Real-time qRT-PCR primers: Igfbp3 | Qiagen | Cat# PPM03820F-200 |
| Real-time qRT-PCR primers:Adam8 | Qiagen | Cat# PPM25830E-200 |
| Real-time qRT-PCR primers:Adam9 | Qiagen | Cat# PPM05228A-200 |
| Real-time qRT-PCR primers:Ccl7 | Qiagen | Cat# PPM02955B-200 |
| Real-time qRT-PCR primers:Ccl8 | Qiagen | Cat# PPM03165A-200 |
| Real-time qRT-PCR primers:Il23a | Qiagen | Cat# PPM03763F-200 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Partek Genomics Suite | Partek | http://www.partek.com/pgs |
| GeneSpring | Agilent Technologies | http://www.agilent.com/en-us/products/software-informatics/life-sciences-informatics/genespring-gx |
| PCR Array Data Analysis | Qiagen | http://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/ |
CONTACT FOR REAGENTS AND RESOURCE SHARING
Request for further information and resources may be directed to Lead Contact Yinling Hu (huy2@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and Human Cell Line and Paraffin Sections
All mice used in this study were cared for in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the National Institutes of Health, and all animal experiments were approved by the IACUC (protocols 11-051, 11-052, 14-051, and 14-052). IkkαKA/KA (Zhu et al., 2007), K5.IKKα (Liu et al., 2011), Ikkαflf and K5.Cre (Liu et al., 2008), Cd4−/− (Stock number 002663, Jackson Laboratory), Igmtm1Cgn (Jackson Laboratory), IkkαAA/AA (Cao et al., 2001), and Rag1−/− mice with a C57BL/6J background were used in this study. All mice were cared at a specific pathogen free (SPF) facility. Mice used in this study were grouped based on simple randomization and genotype without gender preference, and the age was indicated in each experiment correspondingly. Except for IkkαKA/KA mice who have autoimmune issue, all the other mice are normal. The mice were housed in cages with no more than four mice per cage, and housed singly in indicated experiments. All human samples obtained from the Cancer Institute and Cancer Hospital, Chinese Academy of Medical Science, and Peking Union Medical College were approved by this Institute and informed consent has been obtained from patients. The tissue arrays of human esophageal squamous cell carcinomas (SCCs) (ES1202) were purchased from US Biomax, Inc. Human SCC cell lines A431, SCC-25, and NCI-520 were purchased from American Type Culture Collection (ATCC), and were cultured in DMEM, DMEM-F12 Medium, RPMI-1640, respectively, with 10% of fetal bovine serum (FBS) and 1% penicillin streptomycin.
MOTHOD DETAILS
Reagents and Antibodies
Amphotericin B was purchased from X-GEN Pharmaceuticals Inc. Zymosan A was purchased from Sigma-Aldrich (Z4250-250 mg) and was boiled for one hour before use. The YPD agar medium was purchased from Clontech (Clontech, 630410). GW2974 was purchased from Sigma (G0668-50MG). The following antibodies were used for Western blotting and immunohistochemistry: IKKα (IMG-136A) and IκBα (IMG-127A) from IMAGENEX; IKKβ (05-535) from Millipore; lamin B (sc-6216), RelB (sc-48366), LTβR (sc-80167), p65 (sc-372), IκBα (sc-371), p50 (sc-1190), p16 (sc-377412), p53 (sc-6243), p63 (sc-8431), EGFR (sc-03), and IKKα (sc-7183) from Santa Cruz Biotechnology; Phospho-EGFR (2235), p65 (8242) and p100/p52 (4882) from Cell Signaling Technology; β-actin (A-5441) from Sigma; anti-alpha tubulin antibody (ab4074) from abcam; AIRE Alexa Fluor 488 (53-5934-80) and F4/80 (14-4801-81) from eBioscience; CD4 (550280) and IKKγ (559675) from BD Pharmingen; and biotinylated UEA-1 (B-1065) from Vector Laboratories. The anti-CD40 antibody (rat anti-mCD40 agonist, clone Fgk115) was kindly provided by Dr. Dr. Giorgio Trinchieri’s laboratory.
Fungal Isolation and DNA Sequencing
To isolate fungi from mice, oral swabs and mouse esophageal homogenization were performed under sterile conditions. Briefly, each oral swab was suspended in 200 μl of sterile 1 × PBS, and each fresh mouse esophageal specimen was homogenized in 400 μl of sterile 1× PBS and spread to YPD agar plates containing 10μg/ml of chloramphenicol (referred to as YPD/CAM agar plate). The YPD/CAM agar plates were cultured in an incubator at 30°C for at least three days. Large single colonies were picked up and digested in lysis buffer (100 mM Tris-HCL pH8.0, 200 mM NaCl, 2 mM EDTA, 0.2% SDS) with proteinase K (250μg/ml) at 55°C overnight. Fungal DNA was extracted using phenol-chloroform-isoamyl alcohol (Sigma, P2069-100 ml). Semi-nested PCR was applied to amplify fungal DNAs using universal ITS1 and ITS4 primers for primary PCR, and ITS1 and ITS2 primers for secondary PCR (Chang et al., 2001; Ferrer et al., 2001). PCR products were purified using the Zymoclean™ Gel DNA Recovery Kit (Zymo Research, D4008) and were subjected to direct sequencing using ITS1 and ITS2 primers. DNA sequencing data were analyzed using BLAST to identify fungal species.
Human Paraffin Sample DNA Extraction, Fungal DNA Library Construction and Sequencing
Histological slides of human esophageal squamous cell carcinoma were soaked in xylene at room temperature overnight to remove the cover glass and paraffin, and then each specimen was scratched into a sterile clean tube and digested in lysis buffer (100 mM Tris-HCL pH8.0, 200 mM NaCl, 2 mM EDTA, 0.2% SDS) with proteinase K (250 μg/ml) at 55°C overnight. Tissue DNA was extracted using phenol-chloroform-isoamyl alcohol (Sigma, P2069-100 ml). PCR was performed to amplify potential fungal DNA using ITS86F and ITS4 primers. PCR products were used to make fungal DNA libraries using TOPO TA cloning kits (Invitrogen). Colonies containing inserts were sequenced using T3 primer.
Antifungal Treatment
Eight-week-old IkkαKA/KA mice with C57BL/6J background were treated with 100μl of amphotericin B at a 1mg/kg dosage twice a week for three months through intraperitoneal injection. Amphotericin B was dissolved in sterile endotoxin-free water and diluted in sterile 5% dextrose solution. After three months of treatment, mice were euthanized for histologic examinations.
Fungal Infection
Eight-week-old mice were inoculated orally with 30μl of the 2 × 107 organism suspension of Cladosporium cladosporioides each time. The inoculation was performed biweekly, for a total of five times. The oral swab culture was used to monitor the fungal infection after oral inoculation. Three months later, mice were euthanized for histological examinations.
Histopathology, Western Blot Analysis, Immunohistochemical Staining, and Immunofluorescence Staining
The Pathology/Histotechnology Laboratory (PHL) at the Frederick National Laboratory for Cancer Research routinely prepared paraffin sections of human or mouse organs and performed hematoxylin and eosin (H&E) staining and immunohistochemical (IHC) staining of K5, Ki67, CD3, PD-L1, CD68, and F4/80, as well as IKKα for human esophageal tumor tissue array (ES1202, US Biomax), followed by slide scan and data analysis. PHL also performed Grocott’s methenamine silver (GMS) staining and Periodic acid-Schiff (PAS) staining. Eight-μm frozen sections were stained overnight at 4°C with antibodies against K5, UEA-1, and AIRE and were then washed and stained with fluorescence-conjugated secondary antibodies for two hours at room temperature. Finally, the sections were mounted with diamidino-2-phenylindole (DAPI)–mounting medium (Vector Laboratories). Cell lysates (15 μg) or protein extracts from the tissues (20 μg) were resolved on acrylamide gels and examined by Western blotting with specific antibodies, as previously described (Liu et al., 2008).
Isolation of Medullary Thymic Epithelial Cells (mTECs)
Raw mTECs were isolated from the thymuses of six- to eight-week-old mice as described(Lomada et al., 2007). Briefly, the thymuses were finely minced and gently rotated in a 15ml tube in cold RPMI 1640 medium containing 5% FBS at 4°C for three 10-minute intervals to remove the majority of the thymocytes, followed by two washes in cold PBS to remove the serum. The thymic stromal debris was then subjected to either 0.25% trypsin-EDTA (Gibco) digestion at 37°C for one hour for Western blotting sample preparation or liberase TL (Roche) digestion at 37°C for 30 minutes for flow cytometry analysis. The digested TEC suspensions were then filtered using 70μm cell strainers, followed by thymocyte depletion using mouse CD45 MicroBeads (Miltenyi Biotec) (Lomada et al., 2007). Isolated cells were used to prepare cytoplasmic and nuclear proteins or total RNA extraction for further experiments, or stain for flow cytometric analyses.
Ex Vivo T-Cell Responses
Mouse splenocytes were isolated from eight-week-old mice. Six-well plates were coated with 1μg/ml of anti-CD3 antibody (BD, 553057) in sterile 1× PBS overnight at 4°C. Splenocytes were seeded in anti-CD3 antibody-coated six-well plates (4–6 × 106 cells per well) in RPMI 1640 medium containing 10% FBS, 1% glutamine, 50μM 2-ME, and 1% penicillin streptomycin. Splenocytes were then co-cultured with zymosan A (2μg/ml) for three days. After three days of culture, cells were harvested, and quantitative real-time RT-PCR was performed to measure the levels of various cytokines.
Isolation of Macrophages
Peritoneal macrophages were isolated from three- or four-month old mice, and seeded in six-well plates with DMEM-F12 medium containing 10% FBS. After a two-hour incubation period, cells were washed twice to remove cells that were not macrophages.
Flow Cytometry
Mouse thymocytes and splenocytes were isolated in RPMI containing 5% fetal calf serum, followed by red blood cell depletion using the ACK lysing buffer (Gibco). Flow cytometric analysis was performed on cells according to a previously described procedure (Jiang et al., 2011). Antibodies used for analyses: CD3 pacific blue (BD, clone 500A2, 558214), CD8 APC-Cy7 (BD, clone 53-6.7, Cat. No. 557654), CD4 PE (BD, clone GK1.5, Cat. No. 553730), CD80 FITC (BD, Cat. No. 553768), mouse Vβ TCR screen panel (BD, Cat. No. No. 557004), CD25 APC (eBioscience, clone PC61.5, Cat. No. 17-0251-82), Foxp3 FITC (eBioscience, clone FJK16a, Cat. No. 11-5773-82), CD3e FITC (eBioscience, clone 145-2C11, 11-0031-82), CD274 PE (eBioscience, B7-H1, PD-L1, clone MIH5, 12-5982-81), ly-6G (Gr-1) Alexa Fluor 700 (eBioscience, clone RB6-8c5, 56-5931-80), CD11b PE-Cy7 (eBioscience, clone M1/70, 25-0112-81), CD11c 780 (eBioscience, clone N418, 47-0114-80), CD49f (intergrin alpha 6) PE-Cyanine 7 (eBioscience, clone GOH3, Cat. No. 25-0495-80), CD45.1 PE-Cy7 (eBioscience, clone A20, 25-0453-82), CD45.2 eFluor 450 (eBioscience, clone 104, Cat# 48-0454-82), and FITC conjugated UEA1 (Vector, Cat. No. FL-1061). Labeled cells were analyzed on a LSR-II Flow Cytometer (Becton Dickinson). Data were analyzed using FlowJo software.
Preparation of Nuclear Proteins from Cells
Freshly prepared mouse cells were immediately subjected to cytoplasmic and nuclear extraction using NE-PER® Nuclear and Cytoplasmic Extraction Reagents, according to the manufacturer’s instructions (ThermoScientific). Ten μg of cytoplasmic or nuclear lysate were then were used for Western blot analysis.
Total Splenic T-Cell Isolation and Adoptive T-Cell Transfer
Splenocytes were prepared from 10–12-week-old WT and IkkαKA/KA mice. Spleens were mashed using glass slides, subjected to red blood cell lysis using the ACK buffer (Invitrogen), and washed two times with PBS supplemented with 0.2% of BSA and 2 mM of EDTA. T cells were isolated using the Dynabeads Untouched Mouse T Cells Kit (EasySep™ Mouse T Cell Isolation Kit, STEMCELL), and 2 × 106 cells in 0.2 ml of PBS were injected into the tail veins of each mouse at six to seven weeks of age for two injections.
Bone marrow (BM) transfer
Recipient mice were given amoxicillin water for two days prior to and 10 days following irradiation with a lethal dose of 900rad from a cesium 137 source. Five to six hours later, these mice received a 5 x10 6 single suspension of BM cells from donor mice. C57BL/6 IkkαKA/KA mice express CD45.2 (Ly5.2). To study the alternation of donor T cells in chimeric mice, congenic C57BL/6 mice expressing CD45.1 (Ly5.1) (https://www.jax.org/strain/002014) were used as donors or recipients.
Macrophage Depletion
Four-month-old IkkαKA/KA mice on a C57/BL6 background were injected twice per week with 0.1 ml of clodronate-loaded liposomes (The Liposomologists, lipid-based nanoparticles, also known as liposomes, prepared by Encapsula Nano Sciences: www.ClodronateLiposomes.org) for one month through tail intravenous injection (Xiao et al., 2013).
Aspirin and EGFR Inhibitor Treatment
Five-month-old IkkαKA/KA mice were treated with either aspirin water solution or EGFR inhibitor GW2974 suspension added into wet feed food. Each mouse was given 3 mg of aspirin, or 0.6mg of GW2974 per day for one month. Aspirin (A5376-100G: Acetylsalicylic acid) and GW 2974 (G0668) were purchased from Sigma. Mice were singly housed to ensure full consumption of aspirin or GW2974.
Isolation of Macrophages from Mouse Esophagi
The liberase digest stock buffer was prepared by adding 2 ml of serum-free (SF) DMEM medium to one vial of liberase TL (Roche, Cat# 5401119001) on ice for 10–20 minutes. The stock liberase buffer was diluted in 1:10 with SF medium containing 0.2 mg/ml of DNAse I (Sigma, Cat# DN25-1G) on ice. Collected esophagi were placed in a 12-well plate and minced with scissors to small 1–2 mm pieces followed by adding 1 2 ml of digest buffer. The mixtures were incubated at 37°C with mild agitation for 20–40 minutes, separated into single cells via pipet, and passed through cell strainers (Falcon, 70 μM Nylon, Cat# 352350). The collected cells were washed twice with follow cytometry buffer.
ELISA for Autoantibody Detection
ELISA was performed to detect serum autoantibodies binding to cytokines. Wells were coated with carrier-free recombinant mouse IL-17A, IL-17F, and IL-22 (Peprotech) at 1μg/ml in coating buffer (KPL) overnight at 4°C. After blocking, mouse sera diluted 1:20 were incubated for two hours at room temperature before washing and incubation with phosphatase-labeled anti-mouse IgG(γ) (KPL), which was diluted 1:500 for one hour at room temperature, followed by phosphatase substrate (KPL).
RT2 Profiler PCR Array
Total RNAs were extracted using the TRIzol reagent (Ambion, 15596026), and cDNAs were generated using the Tetro cDNA Synthesis Kit (Bioline, BIO-65043). RT2 Profiler PCR arrays were performed using StepOne Plus (Applied Biosystems). SYBR green ROX master mix and primers were used for real-time PCR analysis. The mouse Cytokines & Chemokines RT2 Profiler PCR Array (PAMM-150ZC) was purchased from Qiagen. Data were analyzed according to the Qiagen manual.
Conventional and Quantitative Real-Time (RT)-PCR
Total RNAs were extracted from mouse TECs, total splenic T cells, mouse esophagi, and mouse ESCCs as well. Reverse transcription was performed using the Tetro cDNA Synthesis Kit (Bioline). Quantitative real-time RT-PCR was performed using the StepOnePlus Real-Time PCR System (ABI), and RT-PCR was performed using the MultiGene OptiMax Thermal Cycler (Labnet). The RT-PCR primers used for tissue-restricted genes are: Aire, 5′-GACCTAAACCAGTCCCGGAA-3′ and 5′-ATCCCTTCCACGGCCCCT-3′; Spt1, 5′-GGCTCTGAAACTCAGGCAGA-3′ and 5′-TGCAAACTCATCCACGTTGT-3′; Ccl19, 5′-GCTAATGATGCGGAAGACTG-3′ and 5′-ACTCACATCGACTCTCTAGG-3′; Ccl21, 5′-GCTGCCTTAAGTACAGCCAG-3′ and 5′-GTGTCTGTTCAGTTCTCTTGC-3′; Fabp, 5′-AGACGGAACGGAGCTCAC-3′ and 5′-GCTCTTCAGCGTTGCTCC-3′; Gapdh, 5′-GCAGTGGCAAAGTGGAGATT-3′ and 5′-AGAAGGGGCGGAGATGATGA-3′. RT-PCR used for T-cell repertoire analysis are: Vβ5.1, 5′-AACACTGCCTTCCCTGACCC-3′; Vβ5.2, 5′-GTCTAACACTGTCCTCGCTGATTC-3′; constant region reverse primer, 5′-GCAATCTCTGCTTTTGATGGCT-3′. All the real-time PCR primers were purchased from Qiagen, including Il6 (PPM03015A-200), Tnfα (PPM03113G-200), Ifnγ (PPM03121A-200), Il4 (PPM03013F-200), Il13 (PPM03021B-200), Il17a (PPM03023A-200), Il17f (PPM05398E-200), Il22 (PPM05481A-200), Cxcl11 (PPM03192C-200), Gapdh (PPM02946E-200), Kif2c (Cat No. PPM04983A-200), Cdkn1a (Cat No. PPM02901B-200), Mcm4 (Cat No. PPM03270B-200), Mcm7 (Cat No. PPM03272E-200), Cdk1 (Cat No. PPM02907A-200), Cdk6 (Cat No. PPM02912F-200), Ccne1 (Cat No. PPM02891B-200), Cul3 (Cat No. PPM03257F-200), Setd6 (Cat No. PPM26556B-200), Dnmt1 (Cat No. PPM03685E-200), Pik3cg (Cat No. PPM03469A-200), Igf1 (Cat No. PPM03387F-200), Igfbp3 (Cat No. PPM03820F-200), Adam8 (Cat No. PPM25830E-200), Adam9 (Cat No. PPM05228A-200), Ccl7 (Cat No. PPM02955B-200), Ccl8 (Cat No. PPM03165A-200), and Il23a (Cat No. PPM03763F-200).
Microarray
Total RNAs were isolated from the TECs of five- to six-week-old WT and IkkαKA/KA mice, as well as mouse esophagi and mouse ESCCs using the TRIzol reagent (Invitrogen). RNA quality was examined on a Bioanalyzer and then analyzed on an Affymetrix GeneChip Mouse Genome 430 2.0 array at the Laboratory of Molecular Technology (Cancer Research Technology Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, MD). RNA samples (100 ng of each) were labeled with the Affymetrix IVT Express Labeling Kit (Affymetrix) and hybridized to the Mouse Genome 430 2.0 array according to the manufacturer’s suggested protocol. GeneChips were scanned on an Affymetrix GeneChip Scanner 3000, and data were collected using Affymetrix AGCC software. Microarray data were analyzed using Bioconductor (reference: http://www.ncbi.nlm.nih.gov/pubmed/25633503). Accession numbers of original microarray data (accession number: GSE52137) have been deposited at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). The gene expression profiles of mouse esophageal tissues and esophageal SCCs were analyzed with GeneChip Mouse Genome 430.2 array (affymetrix). The accession number is GSE80005.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters, including the exact value of n that represents number of mice per group, dispersion and precision measures (mean±SEM), and statistical significance are reported in the Figures and Figure Legends. Data is judged to be statistically significant when p <0.05 by selected statistical analysis method, including two-tailed Student’s t test for comparison between two groups, one-way ANOVA for comparison in more than two groups, Fisher’s exact test or Chi-square test for contingency table analysis. In figures, asterisks denote statistical significance as calculated by selected statistical analysis method (*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001). Statistical analysis was performed in GraphPad PRISM 6.
DATA RESOURCES
The microarray analyses for mouse esophageal tissues and tumors (accession number NCBI: GSE80005) and for thymic epithelial cells (accession number NCBI: GSE52137) have been deposited at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/).
Supplementary Material
Acknowledgments
This work was supported by funding from the National Cancer Institute (CA-102510, ZIA BC011212, and ZIA BC 011391) to Y.H.
Footnotes
Supplemental information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online.
AUTHOR CONTRIBUTIONS
Conceptualization, F. Z. and Y.H.; Methodology and Investigation, F.Z., J.W., N.S., D.L., Y.S., L.X., Z.G., Z.Z., S.D., Z.S., and X.W. Writing-Original Draft, F. Z. and Y.H.; Writing-Review & Editing, All Authors; Funding Acquisition, H.Y; Resources, P.Z., Q.Z., and E.R.; Supervision, H.Y.
References
- Akiyama T, Maeda S, Yamane S, Ogino K, Kasai M, Kajiura F, Matsumoto M, Inoue J. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science. 2005;308:248–251. doi: 10.1126/science.1105677. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, Asaumi Y, Kitazawa J, Takayanagi H, Penninger JM, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. doi: 10.1016/j.immuni.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- Beerli RR, Bauer M, Fritzer A, Rosen LB, Buser RB, Hanner M, Maudrich M, Nebenfuehr M, Toepfer JA, Mangold S, et al. Mining the human autoantibody repertoire: isolation of potent IL17A-neutralizing monoclonal antibodies from a patient with thymoma. MAbs. 2014;6:1608–1620. doi: 10.4161/mabs.36292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M. IKKa provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107:763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- Chen L, Deng H, Lu M, Xu B, Wang Q, Jiang J, Wu C. B7-H1 expression associates with tumor invasion and predicts patient’s survival in human esophageal cancer. Intern J Clin Exp Pathol. 2014;7:6015–6023. [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste A, Lagane C, Filipe C, Authier H, Gales A, Bernad J, Douin-Echinard V, Lepert JC, Balard P, Linas MD, et al. IL-13 attenuates gastrointestinal candidiasis in normal and immunodeficient RAG-2(−/−) mice via peroxisome proliferator-activated receptor-gamma activation. J Immunol. 2008;180:4939–4947. doi: 10.4049/jimmunol.180.7.4939. [DOI] [PubMed] [Google Scholar]
- Descargues P, Sil AK, Sano Y, Korchynskyi O, Han G, Owens P, Wang XJ, Karin M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc Natl Acad Sci USA. 2008;105:2487–2492. doi: 10.1073/pnas.0712044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6:e1000713. doi: 10.1371/journal.ppat.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, Reich K, Amato D, Ball SG, Braun DK, et al. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. N Engl J Med. 2016;375:345–356. doi: 10.1056/NEJMoa1512711. [DOI] [PubMed] [Google Scholar]
- Hamazaki Y, Fujita H, Kobayashi T, Choi Y, Scott HS, Matsumoto M, Minato N. Medullary thymic epithelial cells expressing Aire represent a unique lineage derived from cells expressing claudin. Nat Immunol. 2007;8:304–311. doi: 10.1038/ni1438. [DOI] [PubMed] [Google Scholar]
- Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKa subunit of IkB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. IKKa controls formation of the epidermis independently of NF-kB. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- Kogawa K, Kudoh J, Nagafuchi S, Ohga S, Katsuta H, Ishibashi H, Harada M, Hara T, Shimizu N. Distinct clinical phenotype and immunoreactivity in Japanese siblings with autoimmune polyglandular syndrome type 1 (APS-1) associated with compound heterozygous novel AIRE gene mutations. Clin Immunol. 2002;103:277–283. doi: 10.1006/clim.2002.5208. [DOI] [PubMed] [Google Scholar]
- Li L, Ruan Q, Hilliard B, Devirgiliis J, Karin M, Chen YH. Transcriptional regulation of the Th17 immune response by IKK(alpha) J Exp Med. 2011;208:787–796. doi: 10.1084/jem.20091346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Sun XJ, Han QB, Wang Z, Xu YP, Gu JL, Wu W, Zhang GU, Hu JL, Sun WY, et al. Epidermal growth factor receptor protein overexpression and gene amplification are associated with aggressive biological behaviors of esophageal squamous cell carcinoma. Oncol Lett. 2015;10:901–906. doi: 10.3892/ol.2015.3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Park E, Zhu F, Bustos T, Liu J, Shen J, Fischer SM, Hu Y. A critical role for I{kappa}B kinase {alpha} in the development of human and mouse squamous cell carcinomas. Proc Natl Acad Sci USA. 2006;103:17202–17207. doi: 10.1073/pnas.0604481103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Willette-Brown J, Liu S, Chen X, Fischer SM, Hu Y. IKKalpha represses a network of inflammation and proliferation pathways and elevates c-Myc antagonists and differentiation in a dose-dependent manner in the skin. Cell Death Differ. 2011;18:1854–1864. doi: 10.1038/cdd.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Xia X, Zhu F, Park E, Carbajal S, Kiguchi K, DiGiovanni J, Fischer SM, Hu Y. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell. 2008;14:212–225. doi: 10.1016/j.ccr.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomada D, Liu B, Coghlan L, Hu Y, Richie ER. Thymus Medulla Formation and Central Tolerance Are Restored in IKK{alpha}−/− Mice That Express an IKK{alpha} Transgene in Keratin 5+ Thymic Epithelial Cells. J Immunol. 2007;178:829–837. doi: 10.4049/jimmunol.178.2.829. [DOI] [PubMed] [Google Scholar]
- Maeda G, Chiba T, Kawashiri S, Satoh T, Imai K. Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin Cancer Res. 2007;13:5041–5047. doi: 10.1158/1078-0432.CCR-07-0463. [DOI] [PubMed] [Google Scholar]
- Manley NR, Richie ER, Blackburn CC, Condie BG, Sage J. Structure and function of the thymic microenvironment. Front Biosci. 2011;16:2461–2477. doi: 10.2741/3866. [DOI] [PubMed] [Google Scholar]
- Marinari B, Moretti F, Botti E, Giustizieri ML, Descargues P, Giunta A, Stolfi C, Ballaro C, Papoutsaki M, Alema S, et al. The tumor suppressor activity of IKKalpha in stratified epithelia is exerted in part via the TGF-beta antiproliferative pathway. Proc Natl Acad Sci USA. 2008;105:17091–17096. doi: 10.1073/pnas.0809288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis D, Benoist C. Aire. Annu Rev Immunol. 2009;27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532. [DOI] [PubMed] [Google Scholar]
- Mermel LA, Allon M, Bouza E, Craven DE, Flynn P, O’Grady NP, Raad II, Rijnders BJ, Sherertz RJ, Warren DK. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:1–45. doi: 10.1086/599376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi HJ, Laakso S, Salminen JT, Arstila TP, Tuulasvaara A. A normal T cell receptor beta CDR3 length distribution in patients with APECED. Cell Immunol. 2015;295:99–104. doi: 10.1016/j.cellimm.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Ostanin DV, Barlow S, Shukla D, Grisham MB. NADPH oxidase but not myeloperoxidase protects lymphopenic mice from spontaneous infections. Biochem Biophys Res Commun. 2007;355:801–806. doi: 10.1016/j.bbrc.2007.02.029. [DOI] [PubMed] [Google Scholar]
- Park E, Zhu F, Liu B, Xia X, Shen J, Bustos T, Fischer SM, Hu Y. Reduction in IkappaB kinase alpha expression promotes the development of skin papillomas and carcinomas. Cancer Res. 2007;67:9158–9168. doi: 10.1158/0008-5472.CAN-07-0590. [DOI] [PubMed] [Google Scholar]
- Polack FM, Siverio C, Bresky RH. Corneal chromomycosis: double infection by Phialophora verrucosa (Medlar) and Cladosporium cladosporioides (Frescenius) Ann Ophthalmol. 1976;8:139–144. [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kampe O, Eskelin P, Pelto-Huikko M, Peltonen L. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet. 2002;11:397–409. doi: 10.1093/hmg/11.4.397. [DOI] [PubMed] [Google Scholar]
- Rautemaa R, Hietanen J, Niissalo S, Pirinen S, Perheentupa J. Oral and oesophageal squamous cell carcinoma--a complication or component of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, APS-I) Oral Oncol. 2007;43:607–613. doi: 10.1016/j.oraloncology.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Ryan KR, Hong M, Arkwright PD, Gennery AR, Costigan C, Dominguez M, Denning D, McConnell V, Cant AJ, Abinun M, et al. Impaired dendritic cell maturation and cytokine production in patients with chronic mucocutanous candidiasis with or without APECED. Clin Exp Immunol. 2008;154:406–414. doi: 10.1111/j.1365-2249.2008.03778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- Stoner GD, Gupta A. Etiology and chemoprevention of esophageal squamous cell carcinoma. Carcinogenesis. 2001;22:1737–1746. doi: 10.1093/carcin/22.11.1737. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. 2014;14:405–416. doi: 10.1038/nri3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Joosten LA, Shaw PJ, Smeekens SP, Malireddi RK, van der Meer JW, Kullberg BJ, Netea MG, Kanneganti TD. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Europ J Immunol. 2011;41:2260–2268. doi: 10.1002/eji.201041226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Liu S, Xiao Z, Zhu F, Song NY, Zhou M, Liu B, Shen J, Nagashima K, Veenstra TD, et al. An IKKalpha-Nucleophosmin Axis Utilizes Inflammatory Signaling to Promote Genome Integrity. Cell Rep. 2013;5:1243–1255. doi: 10.1016/j.celrep.2013.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Park E, Liu B, Willette-Brown J, Gong W, Wang J, Mitchell D, Fischer SM, Hu Y. Reduction of IKKalpha expression promotes chronic ultraviolet B exposure-induced skin inflammation and carcinogenesis. Am J Pathol. 2010;176:2500–2508. doi: 10.2353/ajpath.2010.091041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Jiang Q, Willette-Brown J, Xi S, Zhu F, Burkett S, Back T, Song NY, Datla M, Sun Z, et al. The Pivotal Role of IKKalpha in the Development of Spontaneous Lung Squamous Cell Carcinomas. Cancer Cell. 2013;23:527–540. doi: 10.1016/j.ccr.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Zhang Y, He B, Xiang J, Wang ZF, Zheng FM, Xu J, Chen MY, Zhu YL, Wen HJ, et al. IKKalpha restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat Commun. 2014;5:3661. doi: 10.1038/ncomms4661. [DOI] [PubMed] [Google Scholar]
- Zhu F, Xia X, Liu B, Shen J, Hu Y, Person M, Hu Y. IKKalpha Shields 14-3-3sigma, a G(2)/M Cell Cycle Checkpoint Gene, from Hypermethylation, Preventing Its Silencing. Mol Cell. 2007;27:214–227. doi: 10.1016/j.molcel.2007.05.042. [DOI] [PubMed] [Google Scholar]
- Zhu W, Phan QT, Boontheung P, Solis NV, Loo JA, Filler SG. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc Natl Acad Sci USA. 2012;109:14194–14199. doi: 10.1073/pnas.1117676109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann KC, Sarbia M, Weber AA, Borchard F, Gabbert HE, Schror K. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res. 1999;59:198–204. [PubMed] [Google Scholar]
- Chang HC, Leaw SN, Huang AH, Wu TL, Chang TC. Rapid identification of yeasts in positive blood cultures by a multiplex PCR method. J Clin Microbiol. 2001;39:3466–3471. doi: 10.1128/JCM.39.10.3466-3471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer C, Colom F, Frases S, Mulet E, Abad JL, Alio JL. Detection and identification of fungal pathogens by PCR and by ITS2 and 5.8S ribosomal DNA typing in ocular infections. J Clin Microbiol. 2001;39:2873–2879. doi: 10.1128/JCM.39.8.2873-2879.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Weiss JM, Back T, Chan T, Ortaldo JR, Guichard S, Wiltrout RH. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res. 2011;71:4074–4084. doi: 10.1158/0008-5472.CAN-10-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein P, Strausbauch M, Therneau T, Borson N. The application of real-time PCR to the analysis of T cell repertoires. Nucleic Acids Res. 2008;36:e140. doi: 10.1093/nar/gkn634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.