

Highlights
-
•
The SARS epidemic drew attention to bats as major coronavirus hosts.
-
•
The known coronavirus genetic diversity is much higher in bats than in any other mammalian host.
-
•
Lack of bat coronavirus isolates and full genomes challenge taxonomic classification.
-
•
Viruses closely related to SARS-CoV, MERS-CoV and HCoV-229E exist in bats.
-
•
Mechanisms of putative host switches from bats into humans are unknown.
Keywords: Coronaviridae, Alphacoronavirus, Betacoronavirus, Bats, Zoonosis, Taxonomy
Abstract
In 2002/2003, a novel coronavirus (CoV) caused a pandemic, infecting more than 8000 people, of whom nearly 10% died. This virus, termed severe acute respiratory syndrome-CoV was linked to a zoonotic origin from rhinolophid bats in 2005. Since then, numerous studies have described novel bat CoVs, including close relatives of the newly emerging Middle East respiratory syndrome (MERS)-CoV. In this paper we discuss CoV genomic properties and compare different taxonomic approaches in light of the technical difficulties of obtaining full genomic sequences directly from bat specimens. We first present an overview of the available studies on bat CoVs, with details on their chiropteran hosts, then comparatively analyze the increase in bat CoV studies and novel genomic sequences obtained since the SARS pandemic. We then conduct a comprehensive phylogenetic analysis of the genera Alpha- and Betacoronavirus, to show that bats harbour more CoV diversity than other mammalian hosts and are widely represented in most, but not all parts of the tree of mammalian CoVs. We next discuss preliminary evidence for phylogenetic co-segregation of CoVs and bat hosts encompassing the Betacoronavirus clades b and d, with an emphasis on the sampling bias that exists among bat species and other mammals, then present examples of CoVs infecting different hosts on the one hand and viruses apparently confined to host genera on the other. We also demonstrate a geographic bias within available studies on bat CoVs, and identify a critical lack of information from biodiversity hotspots in Africa, Asia and Latin America. We then present evidence for a zoonotic origin of four of the six known human CoVs (HCoV), three of which likely involved bats, namely SARS-CoV, MERS-CoV and HCoV-229E; compare the available data on CoV pathogenesis in bats to that in other mammalian hosts; and discuss hypotheses on the putative insect origins of CoV ancestors. Finally, we suggest caution with conclusions on the zoonotic potential of bat viruses, based only on genomic sequence data, and emphasize the need to preserve these ecologically highly relevant animals. This paper forms part of a symposium in Antiviral Research on “from SARS to MERS: 10 years of research on highly pathogenic human coronaviruses”.
1. Introduction
In 2002/2003, an outbreak of severe respiratory disease occurred, infecting 8096 people worldwide and killing 774 (9.5%) of them. A novel human coronavirus (CoV) named severe acute respiratory syndrome (SARS)-CoV was identified as the causative agent (Drosten et al., 2003). This virus was much more pathogenic than previously known human coronaviruses (HCoV) mainly causing mild respiratory disease (van der Hoek, 2007). The clinical aspects of the SARS epidemic and research on the SARS-CoV have been reviewed in two other articles in the present series (Cheng et al., 2013, Hilgenfeld and Peiris, 2013).
Animal CoV have been known since the late 1930s, including viruses that are highly pathogenic for livestock, pet and laboratory animals, such as transmissible gastroenteritis virus of swine (TGEV), bovine CoV (BCoV), feline infectious peritonitis virus (FIPV), mouse hepatitis virus (MHV) and infectious bronchitis virus (IBV) (Saif, 2004). The earliest descriptions of two HCoVs termed HCoV-229E and -OC43 were already made in the 1960s (Hamre and Procknow, 1966, McIntosh et al., 1967). In the SARS aftermath, two additional HCoV termed -NL63 and -HKU1 were detected (van der Hoek et al., 2004, Woo et al., 2005). In 2012, a highly pathogenic sixth HCoV, termed Middle East respiratory syndrome (MERS)-CoV emerged in the Arabian peninsula (de Groot et al., 2013, Zaki et al., 2012, Hilgenfeld and Peiris, 2013).
During the SARS epidemic, first hints pointed to a zoonotic origin of the SARS-CoV, with civets as the suspected source of human infection (Guan et al., 2003, Song et al., 2005, Xu et al., 2004). Genetically diversified CoVs related to SARS-CoV were then found in Chinese rhinolophid bats, indicating these animals may constitute the animal reservoir of this novel HCoV (Lau et al., 2005, Li et al., 2005). These findings and the concomitant description of Ebola viruses in African flying foxes (Leroy et al., 2005) triggered research into bats as hosts of emerging pathogens. Among the factors promoting bats as animal reservoirs for mammalian viruses are their longevity, densely packed colonies, close social interaction and their ability to fly (Calisher et al., 2006, Luis et al., 2013). The numerous descriptions of novel viruses in bats and other animals have drastically changed our perception of the relevance of animal reservoirs for the understanding of emerging zoonoses (Drosten, 2013, Karesh et al., 2012, Morse et al., 2012). In analogy to SARS-CoV, the novel MERS-CoV may share a putative origin in bats (Annan et al., 2013). This review focuses on bats and the CoVs they host.
1.1. Coronavirus genomic organization
CoVs belong to the order Nidovirales, family Coronaviridae and subfamily Coronavirinae, comprising four genera termed Alpha-, Beta-, Gamma- and Deltacoronavirus (Adams and Carstens, 2012, Perlman and Netland, 2009). Betacoronaviruses are further separated into clades a–d, while the separation between alphacoronavirus clades a and b has been discontinued (de Groot et al., 2012).
CoV genomes are composed of one continuous RNA strand of positive polarity, ranging between approximately 27 and 32,000 nucleotides, constituting the largest continuous RNA genomes among mammalian viruses (Woo et al., 2009). Starting from the 5′-end of these genomes, approximate two-thirds encodes a large open reading frame (ORF) 1, producing up to 16 nonstructural proteins (nsp). A conserved sequence UUUAAAC, located around genome positions 12–14,000 in alpha- and betacoronaviruses, provides the characteristic ribosomal slippage leading to transcription of ORF1ab (Brian and Baric, 2005, Masters, 2006). The functions of individual nsp are only partially understood.
Known ORF1ab gene products include, among others, a papain-like protease (Plpro) and main protease (Mpro), a helicase, two methyltransferases, a RNA-dependent RNA polymerase (RdRp) and several innate immunity antagonists (Perlman and Netland, 2009). Downstream of the ORF1ab, all CoVs contain genes coding for the structural proteins spike, envelope, membrane and nucleocapsid and several accessory genes in variable number and genomic location (Woo et al., 2009). A unique CoV feature is the discontinuous transcription of genes downstream of the ORF1ab from subgenomic mRNAs, involving interaction between characteristic transcription-regulatory sequences (TRS) found in the most 5′-genomic region (leader TRS) and upstream of individual genes (body TRS) (Brian and Baric, 2005, Pasternak et al., 2006, Perlman and Netland, 2009, Woo et al., 2009).
1.2. Bat coronavirus detection and challenges for their taxonomic classification
Almost all CoV field studies investigating bats or other animals are based on PCR assays targeting parts of the ORF1ab, typically the RdRp. In these studies, PCR amplicon sizes range from as little as 121 to around 404 base-pairs (Anthony et al., 2013a, da Silva Filho et al., 2012, de Souza Luna et al., 2007, Moes et al., 2005, Tong et al., 2009). It should be noted that these small amplicon sizes drastically hinder CoV phylogenetic reconstructions. Therefore, caution should be taken when drawing conclusions on the relationships within the subfamily Coronavirinae, based on such small sequence fragments.
There are only few characterizations of complete CoV genomes from bat feces, as summarized in Table 1 . The lack of virus isolates obtained directly from bats challenges the complete genomic characterization of these large and highly variable RNA viruses. The information originating from the four available metagenomic studies reporting CoV sequences offers even less phylogenetic power, because the resulting genomic fragments are very variable in genomic location and length (Donaldson et al., 2010, Ge et al., 2012, Li et al., 2010, Wu et al., 2012b).
Table 1.
Studies yielding bat coronavirus sequences.
| Continent | Bat family yielding CoV sequences | Country | Reference | Comments |
|---|---|---|---|---|
| America | Phyllostomidae | Brazil | Brandao et al. (2008) | No full genome information available |
| Molossidae | Brazil | Lima et al. (2013) | ||
| Phyllostomidae, Molossidae | Brazil | Corman et al. (2013b) | ||
| Phyllostomidae, Molossidae, Noctilionidae, Vespertilionidae | Brazil | Góes et al. (2013) | ||
| Vespertilionidae | Canada | Misra et al. (2009) | ||
| Phyllostomidae, Mormoopidae | Costa Rica | Corman et al. (2013b) | ||
| Phyllostomidae, Emballonuridae, Mormoopidae, Vespertilionidae | Mexico | Góes et al. (2013) | ||
| Phyllostomidae, Mormoopidae, Molossidae, Vespertilionidae | Mexico | Anthony et al. (2013a) | ||
| Phyllostomidae | Panama | Corman et al. (2013b) | ||
| Phyllostomidae | Trinidad | Carrington et al. (2008) | ||
| Vespertilionidae | USA | Dominguez et al. (2007) | ||
| Vespertilionidae, Molossidae | USA | Li et al. (2010) | ||
| Vespertilionidae | USA | Donaldson et al. (2010) | ||
| Vespertilionidae | USA | Osborne et al. (2011) | ||
| Vespertilionidae | USA | Huynh et al. (2012) | ||
| Europe | Rhinolophidae, Vespertilionidae | Bulgaria | Drexler et al. (2010) | SARS-related CoV; Full genome |
| Vespertilionidae | UK | August et al. (2012) | No full genome information available | |
| Vespertilionidae | Germany | Gloza-Rausch et al. (2008) | ||
| Vespertilionidae | Germany | Drexler et al. (2010) | ||
| Vespertilionidae | Germany | Drexler et al. (2011) | ||
| Rhinolophidae | Italy | Balboni et al. (2011) | ||
| Rhinolophidae | Italy | Balboni et al. (2012) | ||
| Vespertilionidae | Netherlands | Reusken et al. (2010) | ||
| Vespertilionidae | Netherlands | Annan et al. (2013) | ||
| Vespertilionidae | Roumania | Annan et al. (2013) | ||
| Rhinolophidae | Slovakia | Rihtaric et al. (2010) | ||
| Vespertilionidae | Spain | Falcon et al. (2011) | ||
| Vespertilionidae | Ukraine | Annan et al. (2013) | ||
| Africa | Hipposideridae | Ghana | Pfefferle et al. (2009) | No full genome information available |
| Nycteridae | Ghana | Annan et al. (2013) | ||
| Pteropodidae, Hipposideridae, Vespertilionidae, Molossidae | Kenya | Tong et al. (2009) | ||
| Pteropodidae, Megadermatidae, Vespertilionidae, Molossidae | Kenya | Tao et al. (2012) | ||
| Hipposideridae | Nigeria | Quan et al. (2010) | ||
| Vespertilionidae, Molossidae | South Africa | Geldenhuys et al. (2013) | ||
| Vespertilionidae | South Africa | Ithete et al. (2013) | ||
| Asia | Pteropodidae | Bangladesh | Anthony et al. (2013b) | No full genome information available |
| Vespertilionidae | China | Poon et al. (2005) | ||
| Rhinolophidae, Vespertilionidae | China | Woo et al. (2006) | ||
| Vespertilionidae | China | Chu et al. (2006) | ||
| Pteropodidae | China | Lau et al. (2010b) | ||
| Rhinolophidae | China | Yuen et al. (2012) | ||
| Hipposideridae | China | Ge et al. (2012) | ||
| Rhinolophidae, Vespertilionidae | China | Wu et al. (2012b) | ||
| Vespertilionidae | Japan | Shirato et al. (2012) | ||
| Emballonuridae, Pteropodidae, Rhinopomatidae, Vespertilionidae | Saudi Arabia | Memish et al. (2013) | ||
| Pteropodidae, Vespertilionidae | Philippines | Watanabe et al. (2010) | ||
| Pteropodidae, Hipposideridae | Philippines | Tsuda et al. (2012) | ||
| Hipposideridae | Thailand | Gouilh et al. (2011) | ||
| Molossidae | Thailand | Wacharapluesadee et al. (2013) | ||
| Vespertilionidae | China | Chu et al. (2008) | 1A/1B, HKU8; full genome | |
| Pteropodidae, Hipposideridae | China | Lau et al. (2012) | HKU10; full genomes | |
| Rhinolophidae | China | Lau et al. (2007) | HKU2; full genome | |
| Pteropodidae, Rhinolophidae,Vespertilionidae | China | Woo et al. (2007) | HKU4, HKU5, HKU9; full genome | |
| Rhinolophidae, Vespertilionidae | China | Tang et al. (2006) | SARS-related CoV, 512, HKU4; full genome | |
| Rhinolophidae | China | Lau et al. (2005) | SARS-related CoV; full genome | |
| Rhinolophidae | China | Li et al. (2005) | SARS-related CoV; full genome | |
| Rhinolophidae | China | Ren et al. (2006) | SARS-related CoV; full genome | |
| Rhinolophidae | China | Yuan et al. (2010) | SARS-related CoV; full genome | |
| Rhinolophidae | China | Lau et al., 2010a | SARS-related CoV; full genome | |
| Rhinolophidae, Molossidae | China | Yang et al. (2013) | SARS-related CoV; full genomes |
To provide order in CoV taxonomy, the current proposal of the International Committee for the Taxonomy of Viruses (ICTV) is based on pairwise amino acid distances in seven concatenated partial or complete nsp domains, encompassing about 50% of the CoV genome. In technical analogy to an approach that has been validated for the RNA virus family Picornaviridae (Lauber and Gorbalenya, 2012b), amino acid identity below 90% was found to be discriminatory for the designation of novel CoV species (de Groot et al., 2012). A different criterion, relying on RdRp-grouping units (RGU), also used pairwise amino acid distances, but was restricted to a translated 816 nucleotide RdRp (nsp12) fragment. This was chosen as an amenable approach to account for the partial genomic sequences generated by most PCR-based field studies. It was found that defined alphacoronavirus RGU differed by at least 4.8% and betacoronavirus RGU by at least 6.3% in this RdRp fragment. This allowed a surrogate criterion for species definition in the absence of complete genomic sequences (Drexler et al., 2010, Tao et al., 2012). However, the increase in partial CoV sequences has caused a loss of discriminatory power of these RGU thresholds.
For example, the novel HCoV species MERS-CoV (de Groot et al., 2013, van Boheemen et al., 2012) and the genetically closely related Pipistrellus bat CoVs clearly belong to one RGU (Annan et al., 2013, Reusken et al., 2010). However, these novel bat viruses also differ by only 5.1% from the established CoV species HKU5, implying that they share sufficient RdRp sequence identity to be classifiable within both an RGU defined either by MERS-CoV or by HKU5. Re-analysis of all available RdRp sequence data for the present paper indicated that the 4.8% threshold previously determined for alphacoronaviruses could be maintained. In contrast, a revised lower threshold of at least 5.1% amino acid sequence distance in this RdRp fragment was necessary to accommodate all betacoronavirus species included in the current ICTV proposal (de Groot et al., 2012) and the novel partial CoV sequences.
1.3. Increase in coronavirus genomic sequences
Fig. 1 A shows the increase in bat CoV sequence deposition into GenBank that closely followed the 2002–3 SARS pandemic. An even bigger increase of overall CoV sequence entries can be observed after 2009. This is likely due to both the advent of next-generation sequencing techniques and changed GenBank policies requiring host information from submitters. Fig. 1B shows that the proportion of bat CoV sequences among the 5489 overall CoV entries containing host information is small, compared to those from hosts such as ungulates, birds and carnivores. Possible explanations for this difference are likely the veterinary relevance of livestock and pet CoVs and the usage of prototype viruses as laboratory models leading to sequence entries, e.g., for BCoV, TGEV, FIPV and IBV. Still, the number of bat CoV entries (n = 390) almost equalled that of HCoVs (n = 460). However, the lack of host information for HCoVs probably introduces a bias in this comparison.
Fig. 1.
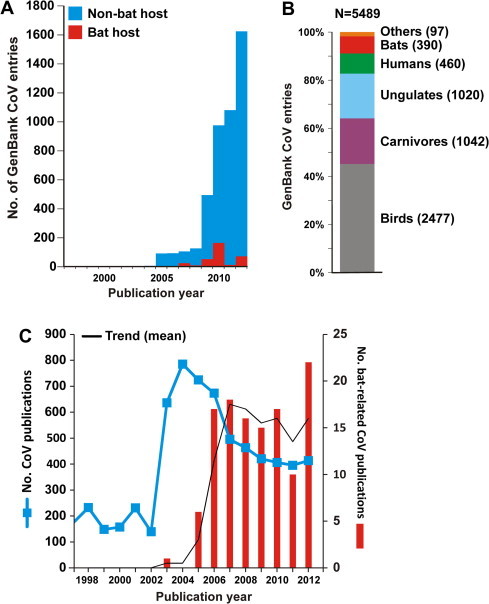
Coronavirus data in public databases. Panels A and B. Data was based on a GenBank search using the terms “Coronaviridae” [ORGANISM] AND Host [All Fields]” in the “Nucleotide” database, on June 16th, 2013. Panel C, PubMed data was retrieved from a search using the terms “coronaviruses, coronavirus, coronaviridae, coronavirinae, bat, chiroptera, bats”, on June 16th, 2013.
An analysis of published CoV research reports is shown in Fig. 1c. The impact of the SARS-epidemic in 2002–3 on the number of CoV publications was tremendous, leading to a near four-fold increase to around 700 published studies per year after 2002. Similarly, the identification of SARS-related CoVs in bats led to around 15 studies on bat CoVs per year after 2005. Of note, this figure also shows that scientific publications began to decrease a few years after SARS-CoV disappeared. The emergence of the MERS-CoV is likely to counteract this phenomenon in the years to come.
1.4. Coronavirus phylogeny
The rapid increase in bat CoV studies enabled hypotheses of bats as reservoir hosts for alpha and betacoronaviruses only a few years after the SARS epidemic (Vijaykrishna et al., 2007, Woo et al., 2009). Fig. 2 shows the clear separation between the mammalian genera Alpha- and Betacoronavirus and the bird-associated genera Gamma- and Deltacoronavirus (Weiss and Navas-Martin, 2005, Woo et al., 2012) in a Bayesian phylogeny based on an 816 RdRp sequence fragment of the complete subfamily Coronavirinae. Fig. 3 provides details of this phylogenetic analysis for the genera Alpha- and Betacoronavirus. The large number of deep branches leading to bat viruses (shown in red) emphasizes the association of these two CoV genera with bat hosts. This is particularly true for the Alphacoronavirus clade formerly designated 1b, which includes HCoV-229E, -NL63 and the Betacoronavirus clades b–d.
Fig. 2.
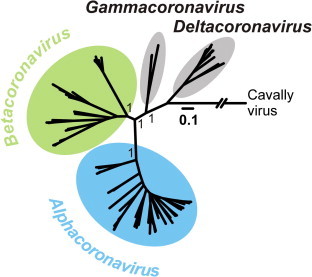
Phylogenetic relationships in the subfamily Coronavirinae. Bayesian phylogeny of an 816-nucleotide RNA-dependent RNA polymerase fragment, as described previously (Drexler et al., 2010) of the subfamily Coronavirinae using MrBayes V3.1 (Ronquist and Huelsenbeck, 2003) under assumption of a GTR + G + I substitution model, using 2,000,000 trees sampled every 100 steps, annotated with a burn-in of 25% using TreeAnnotator V1.7.4 and visualized using FigTree V1.4 from the BEAST package (Drummond et al., 2012). Cavally virus (Zirkel et al., 2011) was used as an outgroup. Values at deep nodes indicate statistical support from Bayesian posterior probabilities, scale bar genetic distance.
Fig. 3.
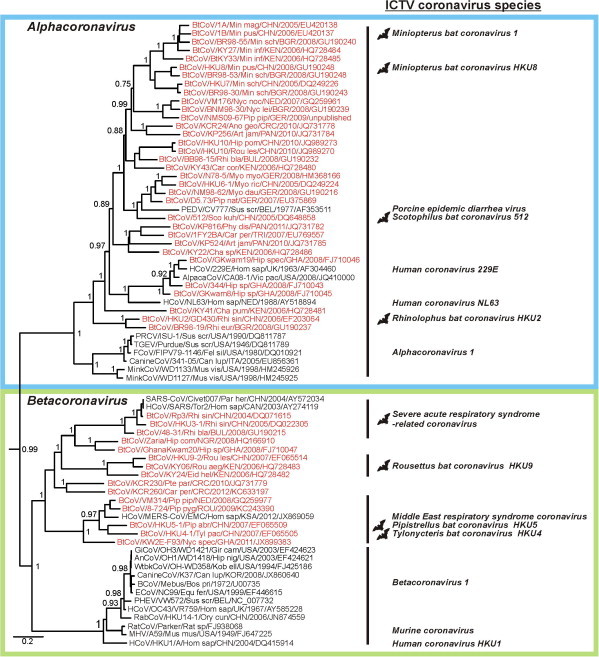
Phylogenetic relationships between coronaviruses and bat hosts. Details of the phylogeny shown in Fig. 2 for the genera Alpha- and Betacoronavirus. ICTV species are given to the right of clade designations and bat symbols, when applicable. Virus designations include strain names, GenBank accession numbers and host information as the first three letters of the latin genus and species names. Bat viruses are shown in red. Boxes indicate Alpha- and Betacoronavirus genera, according to the coloring in Fig. 2.
The relevance of bat CoVs for these genera is also reflected in the current taxonomic proposal of the ICTV (de Groot et al., 2012). Of the 15 recognized species in these two genera, six were only found in bats. These viruses are shown with bat pictograms in Fig. 3 and include Miniopterus bat coronavirus 1, Miniopterus bat coronavirus HKU8, Rhinolophus bat coronavirus HKU2, Scotophilus bat coronavirus 512, Pipistrellus bat coronavirus HKU5, Rousettus bat coronavirus HKU9, and Tylonycteris bat coronavirus HKU4. Many partial bat CoV RdRp sequences were not included in Fig. 3, because only small sequence fragments reducing the phylogenetic resolution were available (Corman et al., 2013b, Drexler et al., 2010). Still, the sequences from these studies (detailed in Table 1) do not alter the overall picture of bat CoV-associated clades within the genera Alpha- and Betacoronavirus.
For most of these bat CoVs, lack of complete genomic sequences prevents their taxonomic designation as species. Still, many of the partial sequences included in Fig. 3 branch deeply in the phylogenetic tree and likely represent not only new species, but even new genetic clades. This is exemplified by the unclassified African Hipposideros betacoronaviruses (Pfefferle et al., 2009, Quan et al., 2010, Tong et al., 2009), which putatively represent a yet to be defined Betacoronavirus clade e and unclassified neotropical Carollia and Pteronotus viruses (Corman et al., 2013b) putatively corresponding to additional Betacoronavirus clades.
Fig. 4 shows that 11 of the 18 extant bat families already contain CoV descriptions, including the two major bat lineages Yinpterochiroptera and Yangochiroptera (Teeling et al., 2005). In most bat families, both alpha- and betacoronaviruses are known, and these detections have originated from both frugivorous and insectivorous bat hosts. Lack of detection in the remaining bat families is likely due to non-exhaustive sampling of the almost 1200 extant bat species (Schipper et al., 2008, Simmons, 2005, Teeling et al., 2005). This void may be filled in future studies.
Fig. 4.
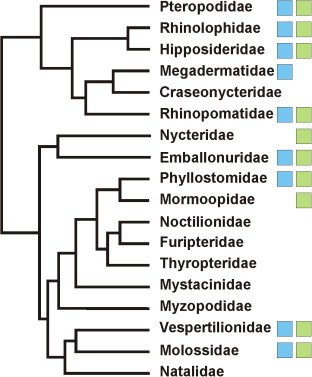
Bat families in which coronaviruses have been detected. Phylogeny of extant bat families is shown according to (Simmons, 2005). Boxes indicate descriptions of alpha- and betacoronaviruses according to the coloring in Fig. 2.
1.5. Bats as coronavirus hosts worldwide
Fig. 5 shows the geographic origin of all 53 studies characterizing novel bat CoVs and Table 1 provides details for these studies. The figure highlights that studies from all continents are now available, but there is a drastic lack of studies from resource-limited or politically unstable settings. Specifically, several biodiversity hotspots linked to the emergence of zoonotic viruses (Jones et al., 2008) are not covered at all, including the Congo basin, large parts of South-East Asia and the Neotropical ecozone. Future sampling of bats from these and other poorly studied areas will likely complete bat species coverage and further increase the known CoV genomic diversity.
Fig. 5.
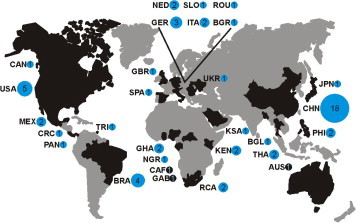
Distribution of bat coronavirus studies. Studies reporting bat CoV sequences by country are indicated, with the number of studies given in blue circles and adjusted in size accordingly. Country codes: BRA = Brazil, CAN = Canada, CRC = Costa Rica, MEX = Mexico, PAN = Panama, TRI = Trinidad and Tobago, USA = United States of America, BGR = Bulgaria, GBR = Great Britain, GER = Germany, ITA = Italy, NED = Netherlands, ROU = Romania, SLO = Slovenia, SPA = Spain, UKR = Ukraine, GHA = Ghana, KEN = Kenia, NGR = Nigeria, RCA = South African Republic, CHN = China, JPN = Japan, PHI = Philippines, THA = Thailand. The black circles for the Central African Republic (CAF), Gabon (GAB) and Australia (AUS) indicate published sequences in GenBank, but lack of publication of the corresponding studies. Countries where CoV studies have been performed are in black, others in grey.
1.6. Association of coronavirus clades with mammalian hosts
1.6.1. Promiscuous versus host-specific coronaviruses
Only a small fraction of the currently known mammalian CoVs originates from primate, ungulate, lagomorph, carnivore and rodent hosts. As shown in Fig. 3, bats outnumber any other mammalian host in terms of virus diversity. Throughout the CoV phylogeny, examples can be found of both “promiscuous” and very host-restricted viruses. The paramount example of a promiscuous CoV is probably Betacoronavirus 1 (the species including BCoV, HCoV-OC43 and related viruses), which has been detected in cows, horses, dogs, humans, waterbucks, deer, antelopes, camels and giraffes worldwide (Alekseev et al., 2008, Guy et al., 2000, Hasoksuz et al., 2007, Jin et al., 2007, Lim et al., 2013, Majhdi et al., 1997, Zhang et al., 1994). Similarly, FIPV, Canine coronavirus (CCoV) and TGEV are now included in a single species termed Alphacoronavirus 1, and MHV and Rat coronavirus together are now termed Murine coronavirus (de Groot et al., 2012). Another example of an apparently promiscuous CoV is the unclassified bat virus HKU10, which has been detected in the bat families Hipposideridae and Pteropodidae (Lau et al., 2012).
Most other CoVs have been confined to single host genera, exemplified by the detection of SARS-related CoVs and several alphacoronaviruses in Rhinolophus, Myotis, Miniopterus, Nyctalus and Carollia bat hosts, including detections of closely related viruses in individual bats separated by thousands of miles (Corman et al., 2013b, Drexler et al., 2010, Tang et al., 2006). Similarly, Hipposideros betacoronaviruses from Thailand, Kenya, Nigeria and Ghana are closely related (Gouilh et al., 2011, Pfefferle et al., 2009, Quan et al., 2010, Tong et al., 2009) and the betacoronavirus HKU9 has been detected in different species of flying foxes in Africa and Asia (Anthony et al., 2013b, Lau et al., 2010b, Tao et al., 2012, Watanabe et al., 2010, Woo et al., 2007). Of note, the detection of both host-specific and -nonspecific mammalian CoVs parallels what can be observed in the avian Coronavirinae genera. For example, infectious bronchitis virus (IBV, genus Gammacoronavirus) has been detected in a wide range of birds, while the recently described deltacoronaviruses appear to be more host-specific (Chu et al., 2011).
1.6.2. Evidence for phylogenetic co-segregation of coronaviruses and bat hosts
Co-segregation of CoVs and their bat hosts is most visible for HKU9, the Hipposideros betacoronaviruses and the SARS-related CoV, compared to Pteropodidae, Hipposideros and Rhinolophus hosts (Fig. 3, Fig. 4). A striking counter-example is the large number of bat alphacoronaviruses that cluster together with the prototype viruses HCoV-229E, -NL63 and PEDV (Fig. 3). Viruses from numerous bat hosts, together with ungulate and human viruses, are contained in this part of the Alphacoronavirus tree, and in contrast to the betacoronaviruses, the designation of clearly separated subclades is challenging. However, more work needs to be done to formally analyze the degree of phylogenetic co-segregation in the Coronavirinae subfamily.
1.7. Coronavirus host switches from bats
1.7.1. Host switches suggested by phylogenetically-related viruses in humans and bats
The most well-studied CoV host switches have probably occurred from bats to humans. The foremost example is the paradigmatic host switch of SARS-CoV from rhinolophid bats into humans or potentially civets (Balboni et al., 2011, Drexler et al., 2010, Lau et al., 2010a, Lau et al., 2005, Li et al., 2005, Ren et al., 2006, Rihtaric et al., 2010, Yang et al., 2013). These human, civet and bat viruses are now officially summarized by the ICTV in one species, termed SARS-related coronavirus (de Groot et al., 2012).
The genomic relatedness of human and bat SARS-related coronaviruses is greatest in the ORF1ab, while a bat ancestor containing the structural proteins of human SARS-CoV has so far not been detected. Bat SARS-related coronaviruses fail to interact with the human SARS-CoV receptor molecule ACE2, possibly associated with small deletions in their receptor-binding domain (RBD), compared to human SARS-CoV (Li, 2013, Ren et al., 2008). In line with these differences, a bat SARS-related coronavirus synthesized by reverse genetics was only infectious in cell culture and mice when the spike gene was exchanged by the human SARS-CoV homologue (Becker et al., 2008). Because the RBD of European rhinolophid bat SARS-related coronaviruses was more related to that of the human SARS-CoV than the RBD from Chinese bat viruses (Drexler et al., 2010), recombination may have played a role in the emergence of the human pathogenic virus.
However, not all rhinolophid bat species have been tested for SARS-related coronaviruses. For example, only 12 of the at least 19 rhinolophid bat species that occur in China have been tested and SARS-related coronavirus sequence information is only available from 5 of these species (Lau et al., 2005, Li et al., 2005, Poon et al., 2005, Tang et al., 2006, Woo et al., 2006, Woo et al., 2007, Yang et al., 2013, Yuan et al., 2010). Therefore, further studies of Rhinolophus species in Africa, Europe and Asia may provide more insight into the ancestral bat viruses that were the source of the emergence of human SARS-CoV.
It should also be mentioned that the Hipposideros betacoronaviruses detected in Africa and Asia are clearly distinct from SARS-related coronaviruses. These viruses can be distinguished by both sequence distance-based taxonomic approaches described above. Additionally, the phylogenetic position and genomic properties of the unclassified Hipposideros betacoronaviruses differ from SARS-related coronaviruses. These genomic properties include chiefly their different viral 3’-genome ends and accessory ORFs downstream from the membrane gene in the Hipposideros CoVs (Pfefferle et al., 2009, Quan et al., 2010).
Bat ancestors were also found for HCoV-229E in African Hipposideros bats (Pfefferle et al., 2009), and a growing body of data indicates that bats worldwide harbour CoVs related to the MERS-CoV (Annan et al., 2013, Anthony et al., 2013a, Ithete et al., 2013, Reusken et al., 2010, Wacharapluesadee et al., 2013, Woo et al., 2007). Unfortunately, no complete genomes are available yet for the putative bat ancestors of HCoV-229E and MERS-CoV.
Putative MERS-CoV bat ancestors have been most consistently found in the bat family Vespertilionidae and the related family Molossidae. These CoVs include European Pipistrellus bat viruses; a virus termed PML/2011 from a South-African Neoromicia bat (Annan et al., 2013, Ithete et al., 2013); CoVs from a Spanish Hypsugo savii and Eptesicus isabellinus (Falcon et al., 2011); sequences from Thai bat guano (Wacharapluesadee et al., 2013); a CoV from a Mexican Nyctinomops bat (Anthony et al., 2013a); and the fully sequenced betacoronavirus clade c prototype bat CoVs HKU4 and HKU5 from China (Woo et al., 2007). Clade c betacoronaviruses from Ghanaian Nycteris bats were more distantly related to the MERS-CoV (Annan et al., 2013). A short 203-nt RdRp sequence fragment 100% identical to the MERS-CoV prototype strain EMC/2012 was described in a single Saudi-Arabian Taphozous perforatus bat, belonging to the family Emballonuridae (Memish et al., 2013). Although the high sequence identity should have facilitated characterization of other CoV genomic regions, no further virus sequence could be obtained from this specimen.
Detection of closely related CoVs in different bat families is rare, but may occur (Lau et al., 2012, Yang et al., 2013). However, even if closely related MERS-CoV bat ancestors existed in both the distantly related bat families Vespertilionidae and Emballonuridae (Fig. 4), the lack of further CoV sequence information and the single detection of a short genome fragment require further confirmation of this finding. Interestingly, in the same study, a 202-nt sequence from a Rhinopoma hardwickii bat was detected which was 100% identical to BCoV and other Betacoronavirus 1 reference strains (Memish et al., 2013). Again, whether clade a betacoronaviruses also exist in bats requires confirmation by detection in more individual bats and more CoV sequence information.
Regarding potential intermediate hosts of MERS-CoV, camels have been shown to have antibodies against this virus at high rates and titers (Perera et al., 2013, Reusken et al., 2013). Conclusions on the passage of putative bat ancestors of MERS-CoV to humans via camels will only be possible upon genomic characterization of the viruses eliciting this strong antibody response.
Finally, no direct bat ancestor of HCoV-NL63 has ever been found, although the phylogenetic clade containing this HCoV is enclosed by bat CoVs. While the recent success in cultivating HCoV-NL63 on immortalized bat cells may hint at some link between bats and this virus (Huynh et al., 2012), the bat viruses described in that and all previous studies are genetically rather distant from HCoV-NL63 (Corman et al., 2013b).
1.7.2. Mechanisms of host switches
The exact mechanisms by which CoVs adapt to new hosts are unclear. Clearly, virus entry and innate immune responses are among the paramount obstacles to be overcome during viral host switching. The high degree of conservation of the MERS-CoV receptor Dipeptidyl peptidase 4 in different hosts is thus a worrying scenario (Muller et al., 2012, Raj et al., 2013). Generally, it is unclear whether direct zoonotic transmission from bats to humans has occurred for any HCoV. Alternative scenarios involve intermediate hosts, such as carnivores or ungulates (Annan et al., 2013, Cotten et al., 2013, Enserink, 2013, Graham and Baric, 2010, Lau et al., 2013, Song et al., 2005, Wu et al., 2012a).
One way to rapidly acquire novel genes that potentially facilitate host switching is recombination between different viruses. A canonical example of recombination in CoVs is FIPV type 2. The prototype strains of this feline virus are recombinants between FIPV type 1 and CCoV in different parts of the ORF1b and Spike genes (Herrewegh et al., 1998). Recombination has also been hypothesized to be involved in the emergence of the SARS-CoV (Graham and Baric, 2010, Hon et al., 2008, Lau et al., 2010a, Yuan et al., 2010) and HCoV-OC43 genotypes (Lau et al., 2011). However, the observed recombination events in SARS-CoV and HCoV-OC43 are restricted to genetically closely related viruses. The exact recombination partners giving rise to SARS-CoV have never been detected.
Another phenomenon putatively associated with CoV adaptation to distinct hosts and cells is exemplified by the variable deletions spanning over 600 nucleotides in the globular S1 domain of the Spike gene that is associated with a change from enteric (TGEV) to respiratory tract (Porcine respiratory coronavirus, PRCV) tropism. Whether the 290-nucleotide deletion downstream from the Spike gene in HCoV-OC43, in comparison to its likely ancestral virus BCoV (Vijgen et al., 2006, Vijgen et al., 2005), represents an adaptation to a human host remains unknown.
1.8. Coronavirus pathology in bats compared to other mammalian hosts
There is little information on the clinical consequences of CoV infections for bat hosts. In humans, HCoVs-NL63, -OC43, -HKU1 and -229E circulate constantly. SARS-CoV disappeared a few years after its epidemic transmission, while the novel MERS-CoV may only be starting to spread (Enserink, 2013). All HCoV cause primarily respiratory symptoms in humans (Hamre and Procknow, 1966, McIntosh et al., 1967, Peiris et al., 2003, van der Hoek, 2007, van der Hoek et al., 2006, Woo et al., 2005), although the MERS-CoV has also been associated with severe renal complications (Zaki et al., 2012). HCoV shedding in feces is not uncommon (Liu et al., 2004), and there are sporadic reports on CoVs in humans with gastroenteritis, e.g., a case report on BCoV in a pediatric patient leading to the tentative designation Human enteric coronavirus (Zhang et al., 1994). However, CoVs are apparently not generally related to gastroenteritis in humans (Esper et al., 2010).
In comparison, the clinical picture in other animals is considerably more variable, ranging from mild respiratory and gastroenteric symptoms to systemic disease with hepatitis, multi-organ failure and death (summarized in Saif, 2004). The high CoV concentrations in bat feces (Annan et al., 2013, Drexler et al., 2011), together with the recovery of coronaviral RNA from the small and large intestine of frugivorous bats (Watanabe et al., 2010) point to replication in the enteric tract. This is compatible both with the use of bat fecal material or intestinal specimens to detect bat CoVs in almost all published studies and with the high CoV RNA concentrations in the lower intestine of naturally infected hedgehogs (Corman et al., 2013a). Although there are no apparent clinical signs of gastroenteritis (Watanabe et al., 2010) or any other disease in bats, it should be noted that bats appear to raise antibodies against their coronaviruses at high rates (Lau et al., 2010b, Lau et al., 2005, Muller et al., 2007, Tsuda et al., 2012). Whether this correlates with the severity of infection remains unknown.
From an ecologic point of view, CoVs seem to rely on massive amplification on a population level during bat reproductive seasonal cycles, potentially associated with fecal-oral transmission (Drexler et al., 2011, Gloza-Rausch et al., 2008, Osborne et al., 2011). Similar phenomena have been observed for other bat-associated viruses, such as the filo-, henipa-, astro- and lyssaviruses (Amman et al., 2012, Drexler et al., 2011, George et al., 2011, Wacharapluesadee et al., 2010). Still, respiratory or vertical CoV transmission cannot be ruled out at this point, mostly due to the necessity of shielding these protected animals from human interference. Hypothetically, one could question whether insectivorous bats that rely on a functional larynx for echolocation of prey could even tolerate respiratory infection. This highly speculative scenario could point to an evolution of bat (corona-) viruses towards infection outside the respiratory tract. Alternatively, it could represent a sample bias due to the rapid death and decay of bats suffering from any respiratory disease that impairs predation, a likely scenario given the very active metabolism of bats, which for example requires insectivorous bats to consume several grams of insects per night (Encarnação and Dietz, 2006).
2. Discussion
Ten years after the SARS epidemic, there is an overwhelming body of evidence highlighting the relevance of bats for the evolution of mammalian CoVs (Vijaykrishna et al., 2007, Woo et al., 2009). From a taxonomic perspective, there is a clear need for the establishment of reliable criteria for these viruses, such as those established for the family Picornaviridae (Lauber and Gorbalenya, 2012b). The challenges observed in attempts to transfer these distance-based approaches to less well characterized virus families such as the Filoviridae (Lauber and Gorbalenya, 2012a) highlight that the current set of CoV taxonomic criteria will likely have to be optimized and thoroughly validated.
Both the existing ICTV and the RGU-based attempts use a similar methodological background and offer more reliable criteria than phylogeny alone. However, both suffer from several shortcomings. Loss of discriminatory sharpness upon the increment in known CoV genetic diversity will likely limit these approaches, although the ICTV criteria may prove more robust, due to the larger genomic fragments incorporated. From a technical point of view, it should be noted that the extension of the RdRp fragment size used for RGU definitions can be challenging when field specimens contain low RNA concentrations (Corman et al., 2013b, Drexler et al., 2010). A similar difficulty can be observed for the ICTV proposal, which requires the characterization of approximately 50% of a typical CoV genome. The more than 20 unclassified CoV species in the current ICTV proposal demonstrate that this task is not easily fulfilled for viruses which have not been grown in culture (de Groot et al., 2012). Furthermore, both approaches do not incorporate any sequence distance-based data from genomic regions encoding the structural proteins. Besides the technical constraints on characterizing these genes that were stated above, this is likely also due to the poor sequence alignments some of these genes entail. The high variability, e.g., of the Spike gene, complicates the establishment of criteria applicable to the complete subfamily. Still, incorporation of some of the more conserved structural genes, such as the Envelope, Membrane or parts of the Spike and Nucleocapsid genes may prove helpful in carrying out this promising genome-based taxonomic approach.
Regarding mammalian CoV hosts, several questions remain to be answered. First, the complete absence of host-specific CoVs in monkeys or apes impedes our understanding of any putative CoV evolution in other primates than humans. The apparently recent host switches responsible for the generation of HCoVs may indicate that even if CoVs exist in non-human primates (NHP), they may not be relevant for the evolution of their human counterparts. However, the current lack of CoV data from NHPs prevents more definite assertions on this topic.
Second, intermediate hosts adapting bat viruses to humans or other mammals have neither been conclusively found for SARS-CoV, nor for any other HCoV. Despite the high seroprevalence of Chinese animal handlers against SARS-CoV, it is not entirely clear whether civets indeed adapted SARS-CoV for infection in humans (Graham and Baric, 2010, Guan et al., 2003). Similarly, the high seroprevalence of camels against MERS-CoV suggests that these animals are a potential source of the initial introduction of this virus into the human population (Reusken et al., 2013). However, the current lack of MERS-CoV genomic sequences from camels prevents definite conclusions on this putative scenario.
Third, many mammalian orders have not been or are only poorly studied. This is exemplified by the single CoV detected in lagomorphs (rabbit coronavirus HKU14), which likely belongs to the Betacoronavirus 1 species, and by Murine Coronavirus (the former MHV), the only known rodent CoV species. Whether CoVs exist in other mammalian hosts, and to what extent these viruses contribute to our understanding of CoV evolution, for now remain obscure.
Finally, the recent description of distantly related nidoviruses in insects has enabled hypotheses on a putative insect origin of Coronaviridae ancestors (Nga et al., 2011, Zirkel et al., 2011). Interestingly, the number of CoV descriptions from insectivorous bats is large, and a clade c betacoronavirus was recently described from insectivorous hedgehogs (Corman et al., 2013a). Insectivorous mammals, such as shrews, moles and hedgehogs from the order Eulipotyphla could thus be specifically investigated to bolster hypotheses on the putative ancient insect origins of the CoVs harbored by insectivorous mammalian hosts.
It has been hypothesized previously that large genome size, frequent recombination and high mutation rate make coronaviruses particularly prone to zoonotic host switching (Woo et al., 2009). These hallmarks require further consideration. First, the large CoV genomes indeed allow for the accommodation of several genes, putatively enhancing virus pathogenicity. For example, the evolutionary origin of the highly variable SARS-CoV accessory proteins is largely unknown, and deletion of these genes strikingly decreases virulence (McBride and Fielding, 2012, Narayanan et al., 2008). Similarly, among CoV genomes, only members of the betacoronavirus clade contain a hemagglutinin-esterase related to that of influenza C viruses (Zeng et al., 2008), compatible with putative inter-viral gene exchange. Still, little is known about how and when CoVs can incorporate novel genes into their genomes. The technical difficulties observed with the usage of CoV-based vectors for heterologous gene expression indicate that, at least in vitro, the introduction of foreign genes into a CoV genome is not an easy task (de Haan et al., 2005, Sola et al., 2003). Additionally, there are many examples of smaller RNA viruses containing numerous accessory genes that enhance virulence, such as HIV-1 at only a third of the typical CoV genome size (Malim and Emerman, 2008).
Second, it should be kept in mind that CoVs possess proofreading enzymes which limit their mutation rates drastically, compared to other RNA viruses (Eckerle et al., 2010, Hanada et al., 2004, Minskaia et al., 2006). CoVs have thus likely evolved a fine balance between necessary mutation rates and RNA proofreading (Denison et al., 2011). Assumptions of high CoV mutation rates are therefore probably only adequate in specific scenarios, such as the recent host switches of SARS- and MERS-CoV.
Third, recombination may certainly occur during CoV replication, and the prototype example of the recombinant origins of FIPV type 2 was discussed above (Herrewegh et al., 1998, Pasternak et al., 2006). It should be noted, however, that other RNA viruses, such as the negative-stranded paramyxoviruses, which probably recombine much less than CoVs, still give rise to a huge number of likely zoonotic host switches from bats and birds into other mammals, at least rivalling the CoVs (Drexler et al., 2012).
The cumulative evidence suggests that at least four HCoVs share a likely zoonotic origin: HCoV-OC43 from bovines and HCoV-229E, SARS- and MERS-CoV from bats. In comparison, only three influenza A viruses (H1N1, H2N2 and H3N2) have so far successfully established circulation in the human population (Palese, 2004). In contrast to CoVs, influenza A viruses cause regular epidemics or even pandemics. Still, the large number of zoonotic HCoVs emphasizes their potential to infect animals other than their original mammalian hosts, and demonstrates the value of continuous screening of humans and wildlife for potentially emerging pathogens. However, it should be kept in mind that sequence detection alone does not warrant premature conclusions as to the zoonotic potential of such viruses (Deeks et al., 2013, Drosten, 2013). Authentic risk assessment strategies would, for example, rely on infection experiments employing isolated or synthetically reconstructed viruses. In the specific case of the CoVs, identification of genomic markers associated with the viral potential to switch hosts or with increased pathogenicity may enable alternative studies. These markers could include viral interferon antagonists and the RBD, both of which could be analyzed in vitro, even in the absence of virus isolates or completely characterized genomes.
Finally, any attempt to eradicate bats or other wildlife in reaction to virus descriptions should be strongly discouraged, as this counteracts the ecological relevance of these animals, e.g., for pollination and pest reduction (Boyles et al., 2011, Kalka et al., 2008).
Acknowledgements
We are grateful to Monika Eschbach-Bludau, Tobias Bleicker and Sebastian Brünink for technical assistance.
References
- Adams M.J., Carstens E.B. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses (2012) Arch. Virol. 2012;157:1411–1422. doi: 10.1007/s00705-012-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseev K.P., Vlasova A.N., Jung K., Hasoksuz M., Zhang X., Halpin R., Wang S., Ghedin E., Spiro D., Saif L.J. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J. Virol. 2008;82:12422–12431. doi: 10.1128/JVI.01586-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amman B.R., Carroll S.A., Reed Z.D., Sealy T.K., Balinandi S., Swanepoel R., Kemp A., Erickson B.R., Comer J.A., Campbell S., Cannon D.L., Khristova M.L., Atimnedi P., Paddock C.D., Crockett R.J., Flietstra T.D., Warfield K.L., Unfer R., Katongole-Mbidde E., Downing R., Tappero J.W., Zaki S.R., Rollin P.E., Ksiazek T.G., Nichol S.T., Towner J.S. Seasonal pulses of Marburg virus circulation in juvenile Rousettus aegyptiacus bats coincide with periods of increased risk of human infection. PLoS Pathog. 2012;8:e1002877. doi: 10.1371/journal.ppat.1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annan A., Baldwin H.J., Corman V.M., Klose S.M., Owusu M., Nkrumah E.E., Badu E.K., Anti P., Agbenyega O., Meyer B., Oppong S., Sarkodie Y.A., Kalko E.K., Lina P.H., Godlevska E.V., Reusken C., Seebens A., Gloza-Rausch F., Vallo P., Tschapka M., Drosten C., Drexler J.F. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013;19:456–459. doi: 10.3201/eid1903.121503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony S., Ojeda-Flores R., Rico-Chavez O., Navarrete-Macias I., Zambrana-Torrelio C., Rostal M.K., Epstein J.H., Tipps T., Liang E., Sanchez-Leon M., Sotomayor-Bonilla J., Aguirre A.A., Avila R., Medellin R.A., Goldstein T., Suzan G., Daszak P., Lipkin W.I. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013 doi: 10.1099/vir.0.049759-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony S.J., Epstein J.H., Murray K.A., Navarrete-Macias I., Zambrana-Torrelio C.M., Solovyov A., Ojeda-Flores R., Arrigo N.C., Islam A., Ali Khan S., Hosseini P., Bogich T.L., Olival K.J., Sanchez-Leon M.D., Karesh W.B., Goldstein T., Luby S.P., Morse S.S., Mazet J.A., Daszak P., Lipkin W.I. A strategy to estimate unknown viral diversity in mammals. MBio. 2013;4 doi: 10.1128/mBio.00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August T.A., Mathews F., Nunn M.A. Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis. 2012;12:530–533. doi: 10.1089/vbz.2011.0829. [DOI] [PubMed] [Google Scholar]
- Balboni A., Gallina L., Palladini A., Prosperi S., Battilani M. A real-time PCR assay for bat SARS-like coronavirus detection and its application to Italian greater horseshoe bat faecal sample surveys. ScientificWorldJournal. 2012;2012:989514. doi: 10.1100/2012/989514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboni A., Palladini A., Bogliani G., Battilani M. Detection of a virus related to betacoronaviruses in Italian greater horseshoe bats. Epidemiol. Infect. 2011;139:216–219. doi: 10.1017/S0950268810001147. [DOI] [PubMed] [Google Scholar]
- Becker M.M., Graham R.L., Donaldson E.F., Rockx B., Sims A.C., Sheahan T., Pickles R.J., Corti D., Johnston R.E., Baric R.S., Denison M.R. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA. 2008;105:19944–19949. doi: 10.1073/pnas.0808116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles J.G., Cryan P.M., McCracken G.F., Kunz T.H. Conservation. Economic importance of bats in agriculture. Science. 2011;332:41–42. doi: 10.1126/science.1201366. [DOI] [PubMed] [Google Scholar]
- Brandao P.E., Scheffer K., Villarreal L.Y., Achkar S., Oliveira Rde N., Fahl Wde O., Castilho J.G., Kotait I., Richtzenhain L.J. A coronavirus detected in the vampire bat Desmodus rotundus. Braz. J. Infect. Dis. 2008;12:466–468. doi: 10.1590/s1413-86702008000600003. [DOI] [PubMed] [Google Scholar]
- Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calisher C.H., Childs J.E., Field H.E., Holmes K.V., Schountz T. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington C.V., Foster J.E., Zhu H.C., Zhang J.X., Smith G.J., Thompson N., Auguste A.J., Ramkissoon V., Adesiyun A.A., Guan Y. Detection and phylogenetic analysis of group 1 coronaviruses in South American bats. Emerg. Infect. Dis. 2008;14:1890–1893. doi: 10.3201/eid1412.080642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng V.C., Chan J.F., To K.K., Yuen K.Y. Clinical management and infection control of SARS: lessons learned. Antiviral Res. 2013;100:407–419. doi: 10.1016/j.antiviral.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.K., Leung C.Y., Gilbert M., Joyner P.H., Ng E.M., Tse T.M., Guan Y., Peiris J.S., Poon L.L. Avian coronavirus in wild aquatic birds. J. Virol. 2011;85:12815–12820. doi: 10.1128/JVI.05838-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.K., Peiris J.S., Chen H., Guan Y., Poon L.L. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 2008;89:1282–1287. doi: 10.1099/vir.0.83605-0. [DOI] [PubMed] [Google Scholar]
- Chu D.K., Poon L.L., Chan K.H., Chen H., Guan Y., Yuen K.Y., Peiris J.S. Coronaviruses in bent-winged bats (Miniopterus spp.) J. Gen. Virol. 2006;87:2461–2466. doi: 10.1099/vir.0.82203-0. [DOI] [PubMed] [Google Scholar]
- Corman V.M., Kallies R., Philipps H., Göpner G., Müller M.A., Eckerle I., Brünink S., Drosten C., Drexler J.F. J. Virol.; 2013. Characterization of a novel betacoronavirus related to MERS-CoV in European hedgehogs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Rasche A., Diallo T.D., Cottontail V.M., Stocker A., Souza B.F., Correa J.I., Carneiro A.J., Franke C.R., Nagy M., Metz M., Knornschild M., Kalko E.K., Ghanem S.J., Morales K.D., Salsamendi E., Spinola M., Herrler G., Voigt C.C., Tschapka M., Drosten C., Drexler J.F. Highly diversified coronaviruses in neotropical bats. J. Gen. Virol. 2013;94:1984–1994. doi: 10.1099/vir.0.054841-0. [DOI] [PubMed] [Google Scholar]
- Cotten M., Lam T.T., Watson S.J., Palser A.L., Petrova V., Grant P., Pybus O.G., Rambaut A., Guan Y., Pillay D., Kellam P., Nastouli E. Full-genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg. Infect. Dis. 2013;19:736–742. doi: 10.3201/eid1905.130057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Filho L.V., Zerbinati R.M., Tateno A.F., Boas L.V., de Almeida M.B., Levi J.E., Drexler J.F., Drosten C., Pannuti C.S. The differential clinical impact of human coronavirus species in children with cystic fibrosis. J. Infect. Dis. 2012;206:384–388. doi: 10.1093/infdis/jis274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A., Galiano M., Gorbalenya A.E., Memish Z., Perlman S., Poon L.L., Snijder E.J., Stephens G.M., Woo P.C., Zaki A.M., Zambon M., Ziebuhr J. Middle East respiratory syndrome coronavirus (MERS-CoV); announcement of the Coronavirus Study Group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R., Enjuanes L., Gorbalenya A.E., Holmes K.V., Perlman S., Poon L., Rottier P.J.M., Talbot P.J., Woo P.C.Y., Ziebuhr J. Family Coronaviridae. In: King A., Adams M., Carstens E.B., Lefkowitz E.J., editors. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press; Amsterdam, Boston: 2012. pp. 806–820. [Google Scholar]
- de Haan C.A., Haijema B.J., Boss D., Heuts F.W., Rottier P.J. Coronaviruses as vectors: stability of foreign gene expression. J. Virol. 2005;79:12742–12751. doi: 10.1128/JVI.79.20.12742-12751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza Luna L.K., Heiser V., Regamey N., Panning M., Drexler J.F., Mulangu S., Poon L., Baumgarte S., Haijema B.J., Kaiser L., Drosten C. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 2007;45:1049–1052. doi: 10.1128/JCM.02426-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks S., Drosten C., Picker L., Subbarao K., Suzich J. Roadblocks to translational challenges on viral pathogenesis. Nat. Med. 2013;19:30–34. doi: 10.1038/nm.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011;8:270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S.R., O’Shea T.J., Oko L.M., Holmes K.V. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect. Dis. 2007;13:1295–1300. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson E.F., Haskew A.N., Gates J.E., Huynh J., Moore C.J., Frieman M.B. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J. Virol. 2010;84:13004–13018. doi: 10.1128/JVI.01255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J.F., Corman V.M., Muller M.A., Maganga G.D., Vallo P., Binger T., Gloza-Rausch F., Rasche A., Yordanov S., Seebens A., Oppong S., Adu Sarkodie Y., Pongombo C., Lukashev A.N., Schmidt-Chanasit J., Stocker A., Carneiro A.J., Erbar S., Maisner A., Fronhoffs F., Buettner R., Kalko E.K., Kruppa T., Franke C.R., Kallies R., Yandoko E.R., Herrler G., Reusken C., Hassanin A., Kruger D.H., Matthee S., Ulrich R.G., Leroy E.M., Drosten C. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J.F., Corman V.M., Wegner T., Tateno A.F., Zerbinati R.M., Gloza-Rausch F., Seebens A., Muller M.A., Drosten C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011;17:449–456. doi: 10.3201/eid1703.100526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J.F., Gloza-Rausch F., Glende J., Corman V.M., Muth D., Goettsche M., Seebens A., Niedrig M., Pfefferle S., Yordanov S., Zhelyazkov L., Hermanns U., Vallo P., Lukashev A., Muller M.A., Deng H., Herrler G., Drosten C. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010;84:11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C. Virus ecology: a gap between detection and prediction. Emerg. Microbes Infect. 2013;2:e31. doi: 10.1038/emi.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Suchard M.A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle L.D., Becker M.M., Halpin R.A., Li K., Venter E., Lu X., Scherbakova S., Graham R.L., Baric R.S., Stockwell T.B., Spiro D.J., Denison M.R. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encarnação J., Dietz M. Estimation of food intake and ingested energy in Daubenton’s bats (Myotis daubentonii) during pregnancy and spermatogenesis. Eur. J. Wildl. Res. 2006;52:221–227. [Google Scholar]
- Enserink M. Emerging diseases. New coronavirus reveals some of its secrets. Science. 2013;340:17–18. doi: 10.1126/science.340.6128.17. [DOI] [PubMed] [Google Scholar]
- Esper F., Ou Z., Huang Y.T. Human coronaviruses are uncommon in patients with gastrointestinal illness. J. Clin. Virol. 2010;48:131–133. doi: 10.1016/j.jcv.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon A., Vazquez-Moron S., Casas I., Aznar C., Ruiz G., Pozo F., Perez-Brena P., Juste J., Ibanez C., Garin I., Aihartza J., Echevarria J.E. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol. 2011;156:1883–1890. doi: 10.1007/s00705-011-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X., Li Y., Yang X., Zhang H., Zhou P., Zhang Y., Shi Z. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 2012;86:4620–4630. doi: 10.1128/JVI.06671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldenhuys M., Weyer J., Nel L.H., Markotter W. Coronaviruses in South African Bats. Vector Borne Zoonotic Dis. 2013;13:516–519. doi: 10.1089/vbz.2012.1101. [DOI] [PubMed] [Google Scholar]
- George D.B., Webb C.T., Farnsworth M.L., O’Shea T.J., Bowen R.A., Smith D.L., Stanley T.R., Ellison L.E., Rupprecht C.E. Host and viral ecology determine bat rabies seasonality and maintenance. Proc. Natl. Acad. Sci. USA. 2011;108:10208–10213. doi: 10.1073/pnas.1010875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloza-Rausch F., Ipsen A., Seebens A., Gottsche M., Panning M., Drexler J.F., Petersen N., Annan A., Grywna K., Muller M., Pfefferle S., Drosten C. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 2008;14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Góes, L.G.B., Ruvalcaba, S.G., Campos, A.A., Queiroz, L.H., de Carvalho, C., Jerez, J.A., Durigon, E.L., Iñiguez Dávalos, L.I., Dominguez, S.R., 2013. Novel bat coronaviruses, Brazil and Mexico. Emerg. Infect. Dis. epub ahead of print. [DOI] [PMC free article] [PubMed]
- Gouilh M.A., Puechmaille S.J., Gonzalez J.P., Teeling E., Kittayapong P., Manuguerra J.C. SARS-Coronavirus ancestor’s foot-prints in South-East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 2011;11:1690–1702. doi: 10.1016/j.meegid.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Baric R.S. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 2010;84:3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Guy J.S., Breslin J.J., Breuhaus B., Vivrette S., Smith L.G. Characterization of a coronavirus isolated from a diarrheic foal. J. Clin. Microbiol. 2000;38:4523–4526. doi: 10.1128/jcm.38.12.4523-4526.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamre D., Procknow J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 1966;121:190–193. doi: 10.3181/00379727-121-30734. [DOI] [PubMed] [Google Scholar]
- Hanada K., Suzuki Y., Gojobori T. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol. Biol. Evol. 2004;21:1074–1080. doi: 10.1093/molbev/msh109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasoksuz M., Alekseev K., Vlasova A., Zhang X., Spiro D., Halpin R., Wang S., Ghedin E., Saif L.J. Biologic, antigenic, and full-length genomic characterization of a bovine-like coronavirus isolated from a giraffe. J. Virol. 2007;81:4981–4990. doi: 10.1128/JVI.02361-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., Smeenk I., Horzinek M.C., Rottier P.J., de Groot R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998;72:4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R., Peiris J.S.M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon C.C., Lam T.Y., Shi Z.L., Drummond A.J., Yip C.W., Zeng F., Lam P.Y., Leung F.C. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 2008;82:1819–1826. doi: 10.1128/JVI.01926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh J., Li S., Yount B., Smith A., Sturges L., Olsen J.C., Nagel J., Johnson J.B., Agnihothram S., Gates J.E., Frieman M.B., Baric R.S., Donaldson E.F. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 2012;86:12816–12825. doi: 10.1128/JVI.00906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ithete N.L., Stoffberg S., Corman V.M., Cottontail V.M., Richards L.R., Schoeman M.C., Drosten C., Drexler J.F., Preiser W. Close Relative of Human Middle East Respiratory Syndrome Coronavirus in South African Bat. Emerg. Infect. Dis. 2013;19:1697–1699. doi: 10.3201/eid1910.130946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L., Cebra C.K., Baker R.J., Mattson D.E., Cohen S.A., Alvarado D.E., Rohrmann G.F. Analysis of the genome sequence of an alpaca coronavirus. Virology. 2007;365:198–203. doi: 10.1016/j.virol.2007.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.E., Patel N.G., Levy M.A., Storeygard A., Balk D., Gittleman J.L., Daszak P. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalka M.B., Smith A.R., Kalko E.K. Bats limit arthropods and herbivory in a tropical forest. Science. 2008;320:71. doi: 10.1126/science.1153352. [DOI] [PubMed] [Google Scholar]
- Karesh W.B., Dobson A., Lloyd-Smith J.O., Lubroth J., Dixon M.A., Bennett M., Aldrich S., Harrington T., Formenty P., Loh E.H., Machalaba C.C., Thomas M.J., Heymann D.L. Ecology of zoonoses: natural and unnatural histories. Lancet. 2012;380:1936–1945. doi: 10.1016/S0140-6736(12)61678-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Lee P., Tsang A.K., Yip C.C., Tse H., Lee R.A., So L.Y., Lau Y.L., Chan K.H., Woo P.C., Yuen K.Y. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011;85:11325–11337. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Li K.S., Huang Y., Shek C.T., Tse H., Wang M., Choi G.K., Xu H., Lam C.S., Guo R., Chan K.H., Zheng B.J., Woo P.C., Yuen K.Y. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010;84:2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Li K.S., Tsang A.K., Lam C.S., Ahmed S., Chen H., Chan K.H., Woo P.C., Yuen K.Y. Genetic characterization of Betacoronavirus lineage C viruses in bats revealed marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications on the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 2013;87:8638–8650. doi: 10.1128/JVI.01055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Li K.S., Tsang A.K., Shek C.T., Wang M., Choi G.K., Guo R., Wong B.H., Poon R.W., Lam C.S., Wang S.Y., Fan R.Y., Chan K.H., Zheng B.J., Woo P.C., Yuen K.Y. Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault’s rousettes to pomona leaf-nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 2012;86:11906–11918. doi: 10.1128/JVI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Poon R.W., Wong B.H., Wang M., Huang Y., Xu H., Guo R., Li K.S., Gao K., Chan K.H., Zheng B.J., Woo P.C., Yuen K.Y. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J. Virol. 2010;84:11385–11394. doi: 10.1128/JVI.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lau S.K., Woo P.C., Li K.S., Huang Y., Tsoi H.W., Wong B.H., Wong S.S., Leung S.Y., Chan K.H., Yuen K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Nat. Acad. Sci. USA. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Woo P.C., Li K.S., Huang Y., Wang M., Lam C.S., Xu H., Guo R., Chan K.H., Zheng B.J., Yuen K.Y. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology. 2007;367:428–439. doi: 10.1016/j.virol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber C., Gorbalenya A.E. Genetics-based classification of filoviruses calls for expanded sampling of genomic sequences. Viruses. 2012;4:1425–1437. doi: 10.3390/v4091425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber C., Gorbalenya A.E. Toward genetics-based virus taxonomy: comparative analysis of a genetics-based classification and the taxonomy of picornaviruses. J. Virol. 2012;86:3905–3915. doi: 10.1128/JVI.07174-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E.M., Kumulungui B., Pourrut X., Rouquet P., Hassanin A., Yaba P., Delicat A., Paweska J.T., Gonzalez J.P., Swanepoel R. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Res. 2013;100:246–254. doi: 10.1016/j.antiviral.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Victoria J.G., Wang C., Jones M., Fellers G.M., Kunz T.H., Delwart E. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010;84:6955–6965. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Bats are natural reservoirs of SARS-like coronaviruses. Science (New York, NY) 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Lim S.I., Choi S., Lim J.A., Jeoung H.Y., Song J.Y., Dela Pena R.C., An D.J. Complete genome analysis of canine respiratory coronavirus. Genome Announc. 2013;1 doi: 10.1128/genomeA.00093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima F.E., Campos F.S., Kunert Filho H.C., Batista H.B., Carnielli Junior P., Cibulski S.P., Spilki F.R., Roehe P.M., Franco A.C. Detection of Alphacoronavirus in velvety free-tailed bats (Molossus molossus) and Brazilian free-tailed bats (Tadarida brasiliensis) from urban area of Southern Brazil. Virus Genes. 2013;47:164–167. doi: 10.1007/s11262-013-0899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Tang F., Fontanet A., Zhan L., Zhao Q.M., Zhang P.H., Wu X.M., Zuo S.Q., Baril L., Vabret A., Xin Z.T., Shao Y.M., Yang H., Cao W.C. Long-term SARS coronavirus excretion from patient cohort, China. Emerg. Infect. Dis. 2004;10:1841–1843. doi: 10.3201/eid1010.040297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis A.D., Hayman D.T.S., O’Shea T.J., Cryan P.M., Gilbert A.T., Pulliam J.R.C., Mills J.N., Timonin M.E., Willis C.K.R., Cunningham A.A., Fooks A.R., Rupprecht C.E., Wood J.L.N., Webb C.T. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. R. Soc. B: Biol. Sci. 2013;280 doi: 10.1098/rspb.2012.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majhdi F., Minocha H.C., Kapil S. Isolation and characterization of a coronavirus from elk calves with diarrhea. J. Clin. Microbiol. 1997;35:2937–2942. doi: 10.1128/jcm.35.11.2937-2942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim M.H., Emerman M. HIV-1 accessory proteins – ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride R., Fielding B.C. The role of severe acute respiratory syndrome (SARS)-coronavirus accessory proteins in virus pathogenesis. Viruses. 2012;4:2902–2923. doi: 10.3390/v4112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K., Dees J.H., Becker W.B., Kapikian A.Z., Chanock R.M. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc. Nat. Acad. Sci. USA. 1967;57:933–940. doi: 10.1073/pnas.57.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memish, Z.A., Mishra, N., Olival, K.J., Fagbo, S.F., Kapoor, V., Epstein, J.H., AlHakeem, R., Durosinloun, A., Al Asmari, M., Islam, A., Kapoor, A., Briese, T., Daszak, P., Al Rabeeah, A.A., Lipkin, W.I., 2013. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Minskaia E., Hertzig T., Gorbalenya A.E., Campanacci V., Cambillau C., Canard B., Ziebuhr J. Discovery of an RNA virus 3′ → 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA. 2006;103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra V., Dumonceaux T., Dubois J., Willis C., Nadin-Davis S., Severini A., Wandeler A., Lindsay R., Artsob H. Detection of polyoma and corona viruses in bats of Canada. J. Gen. Virol. 2009;90:2015–2022. doi: 10.1099/vir.0.010694-0. [DOI] [PubMed] [Google Scholar]
- Moes E., Vijgen L., Keyaerts E., Zlateva K., Li S., Maes P., Pyrc K., Berkhout B., van der Hoek L., Van Ranst M. A novel pancoronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect. Dis. 2005;5:6. doi: 10.1186/1471-2334-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse S.S., Mazet J.A., Woolhouse M., Parrish C.R., Carroll D., Karesh W.B., Zambrana-Torrelio C., Lipkin W.I., Daszak P. Prediction and prevention of the next pandemic zoonosis. Lancet. 2012;380:1956–1965. doi: 10.1016/S0140-6736(12)61684-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M.A., Paweska J.T., Leman P.A., Drosten C., Grywna K., Kemp A., Braack L., Sonnenberg K., Niedrig M., Swanepoel R. Coronavirus antibodies in African bat species. Emerg. Infect. Dis. 2007;13:1367–1370. doi: 10.3201/eid1309.070342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M.A., Raj V.S., Muth D., Meyer B., Kallies S., Smits S.L., Wollny R., Bestebroer T.M., Specht S., Suliman T., Zimmermann K., Binger T., Eckerle I., Tschapka M., Zaki A.M., Osterhaus A.D., Fouchier R.A., Haagmans B.L., Drosten C. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. MBio. 2012;3 doi: 10.1128/mBio.00515-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Huang C., Makino S. SARS coronavirus accessory proteins. Virus Res. 2008;133:113–121. doi: 10.1016/j.virusres.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nga P.T., Parquet Mdel C., Lauber C., Parida M., Nabeshima T., Yu F., Thuy N.T., Inoue S., Ito T., Okamoto K., Ichinose A., Snijder E.J., Morita K., Gorbalenya A.E. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. 2011;7:e1002215. doi: 10.1371/journal.ppat.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne C., Cryan P.M., O’Shea T.J., Oko L.M., Ndaluka C., Calisher C.H., Berglund A.D., Klavetter M.L., Bowen R.A., Holmes K.V., Dominguez S.R. Alphacoronaviruses in New World bats: prevalence, persistence, phylogeny, and potential for interaction with humans. PLoS ONE. 2011;6:e19156. doi: 10.1371/journal.pone.0019156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P. Influenza: old and new threats. Nat. Med. 2004;10:S82–S87. doi: 10.1038/nm1141. [DOI] [PubMed] [Google Scholar]
- Pasternak A.O., Spaan W.J., Snijder E.J. Nidovirus transcription: how to make sense? J. Gen. Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera R.A., Wang P., Gomaa M.R., El-Shesheny R., Kandeil A., Bagato O., Siu L.Y., Shehata M.M., Kayed A.S., Moatasim Y., Li M., Poon L.L., Guan Y., Webby R.J., Ali M.A., Peiris J.S., Kayali G. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Eurosurveillance. 2013;18:8–14. doi: 10.2807/1560-7917.es2013.18.36.20574. [DOI] [PubMed] [Google Scholar]
- Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S., Oppong S., Drexler J.F., Gloza-Rausch F., Ipsen A., Seebens A., Muller M.A., Annan A., Vallo P., Adu-Sarkodie Y., Kruppa T.F., Drosten C. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009;15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon L.L., Chu D.K., Chan K.H., Wong O.K., Ellis T.M., Leung Y.H., Lau S.K., Woo P.C., Suen K.Y., Yuen K.Y., Guan Y., Peiris J.S. Identification of a novel coronavirus in bats. J. Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan P.L., Firth C., Street C., Henriquez J.A., Petrosov A., Tashmukhamedova A., Hutchison S.K., Egholm M., Osinubi M.O., Niezgoda M., Ogunkoya A.B., Briese T., Rupprecht C.E., Lipkin W.I. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio. 2010;1 doi: 10.1128/mBio.00208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj V.S., Mou H.H., Smits S.L., Dekkers D.H.W., Muller M.A., Dijkman R., Muth D., Demmers J.A.A., Zaki A., Fouchier R.A.M., Thiel V., Drosten C., Rottier P.J.M., Osterhaus A.D.M.E., Bosch B.J., Haagmans B.L. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W., Li W., Yu M., Hao P., Zhang Y., Zhou P., Zhang S., Zhao G., Zhong Y., Wang S., Wang L.F., Shi Z. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J. Gen. Virol. 2006;87:3355–3359. doi: 10.1099/vir.0.82220-0. [DOI] [PubMed] [Google Scholar]
- Ren W., Qu X., Li W., Han Z., Yu M., Zhou P., Zhang S.Y., Wang L.F., Deng H., Shi Z. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 2008;82:1899–1907. doi: 10.1128/JVI.01085-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken C.B., Haagmans B.L., Muller M.A., Gutierrez C., Godeke G.J., Meyer B., Muth D., Raj V.S., Vries L.S., Corman V.M., Drexler J.F., Smits S.L., El Tahir Y.E., De Sousa R., van Beek J., Nowotny N., van Maanen K., Hidalgo-Hermoso E., Bosch B.J., Rottier P., Osterhaus A., Gortazar-Schmidt C., Drosten C., Koopmans M.P. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013;13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken C.B., Lina P.H., Pielaat A., de Vries A., Dam-Deisz C., Adema J., Drexler J.F., Drosten C., Kooi E.A. Circulation of group 2 coronaviruses in a bat species common to urban areas in Western Europe. Vector Borne Zoonotic Dis. 2010;10:785–791. doi: 10.1089/vbz.2009.0173. [DOI] [PubMed] [Google Scholar]
- Rihtaric D., Hostnik P., Steyer A., Grom J., Toplak I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 2010;155:507–514. doi: 10.1007/s00705-010-0612-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Saif L.J. Animal coronaviruses: what can they teach us about the severe acute respiratory syndrome? Rev. Sci. Tech. 2004;23:643–660. doi: 10.20506/rst.23.2.1513. [DOI] [PubMed] [Google Scholar]
- Schipper J., Chanson J.S., Chiozza F., Cox N.A., Hoffmann M., Katariya V., Lamoreux J., Rodrigues A.S., Stuart S.N., Temple H.J., Baillie J., Boitani L., Lacher T.E., Jr., Mittermeier R.A., Smith A.T., Absolon D., Aguiar J.M., Amori G., Bakkour N., Baldi R., Berridge R.J., Bielby J., Black P.A., Blanc J.J., Brooks T.M., Burton J.A., Butynski T.M., Catullo G., Chapman R., Cokeliss Z., Collen B., Conroy J., Cooke J.G., da Fonseca G.A., Derocher A.E., Dublin H.T., Duckworth J.W., Emmons L., Emslie R.H., Festa-Bianchet M., Foster M., Foster S., Garshelis D.L., Gates C., Gimenez-Dixon M., Gonzalez S., Gonzalez-Maya J.F., Good T.C., Hammerson G., Hammond P.S., Happold D., Happold M., Hare J., Harris R.B., Hawkins C.E., Haywood M., Heaney L.R., Hedges S., Helgen K.M., Hilton-Taylor C., Hussain S.A., Ishii N., Jefferson T.A., Jenkins R.K., Johnston C.H., Keith M., Kingdon J., Knox D.H., Kovacs K.M., Langhammer P., Leus K., Lewison R., Lichtenstein G., Lowry L.F., Macavoy Z., Mace G.M., Mallon D.P., Masi M., McKnight M.W., Medellin R.A., Medici P., Mills G., Moehlman P.D., Molur S., Mora A., Nowell K., Oates J.F., Olech W., Oliver W.R., Oprea M., Patterson B.D., Perrin W.F., Polidoro B.A., Pollock C., Powel A., Protas Y., Racey P., Ragle J., Ramani P., Rathbun G., Reeves R.R., Reilly S.B., Reynolds J.E., 3rd, Rondinini C., Rosell-Ambal R.G., Rulli M., Rylands A.B., Savini S., Schank C.J., Sechrest W., Self-Sullivan C., Shoemaker A., Sillero-Zubiri C., De Silva N., Smith D.E., Srinivasulu C., Stephenson P.J., van Strien N., Talukdar B.K., Taylor B.L., Timmins R., Tirira D.G., Tognelli M.F., Tsytsulina K., Veiga L.M., Vie J.C., Williamson E.A., Wyatt S.A., Xie Y., Young B.E. The status of the world’s land and marine mammals: diversity, threat, and knowledge. Science. 2008;322:225–230. doi: 10.1126/science.1165115. [DOI] [PubMed] [Google Scholar]
- Shirato K., Maeda K., Tsuda S., Suzuki K., Watanabe S., Shimoda H., Ueda N., Iha K., Taniguchi S., Kyuwa S., Endoh D., Matsuyama S., Kurane I., Saijo M., Morikawa S., Yoshikawa Y., Akashi H., Mizutani T. Detection of bat coronaviruses from Miniopterus fuliginosus in Japan. Virus Genes. 2012;44:40–44. doi: 10.1007/s11262-011-0661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons N.B. Evolution. An Eocene big bang for bats. Science. 2005;307:527–528. doi: 10.1126/science.1108871. [DOI] [PubMed] [Google Scholar]
- Sola I., Alonso S., Zuniga S., Balasch M., Plana-Duran J., Enjuanes L. Engineering the transmissible gastroenteritis virus genome as an expression vector inducing lactogenic immunity. J. Virol. 2003;77:4357–4369. doi: 10.1128/JVI.77.7.4357-4369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.D., Tu C.C., Zhang G.W., Wang S.Y., Zheng K., Lei L.C., Chen Q.X., Gao Y.W., Zhou H.Q., Xiang H., Zheng H.J., Chern S.W., Cheng F., Pan C.M., Xuan H., Chen S.J., Luo H.M., Zhou D.H., Liu Y.F., He J.F., Qin P.Z., Li L.H., Ren Y.Q., Liang W.J., Yu Y.D., Anderson L., Wang M., Xu R.H., Wu X.W., Zheng H.Y., Chen J.D., Liang G., Gao Y., Liao M., Fang L., Jiang L.Y., Li H., Chen F., Di B., He L.J., Lin J.Y., Tong S., Kong X., Du L., Hao P., Tang H., Bernini A., Yu X.J., Spiga O., Guo Z.M., Pan H.Y., He W.Z., Manuguerra J.C., Fontanet A., Danchin A., Niccolai N., Li Y.X., Wu C.I., Zhao G.P. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA. 2005;102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.C., Zhang J.X., Zhang S.Y., Wang P., Fan X.H., Li L.F., Li G., Dong B.Q., Liu W., Cheung C.L., Xu K.M., Song W.J., Vijaykrishna D., Poon L.L., Peiris J.S., Smith G.J., Chen H., Guan Y. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 2006;80:7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Tang K., Shi M., Conrardy C., Li K.S., Lau S.K., Anderson L.J., Tong S. Genomic characterization of seven distinct bat coronaviruses in Kenya. Virus Res. 2012;167:67–73. doi: 10.1016/j.virusres.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling E.C., Springer M.S., Madsen O., Bates P., O’Brien S.J., Murphy W.J. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307:580–584. doi: 10.1126/science.1105113. [DOI] [PubMed] [Google Scholar]
- Tong S., Conrardy C., Ruone S., Kuzmin I.V., Guo X., Tao Y., Niezgoda M., Haynes L., Agwanda B., Breiman R.F., Anderson L.J., Rupprecht C.E. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda S., Watanabe S., Masangkay J.S., Mizutani T., Alviola P., Ueda N., Iha K., Taniguchi S., Fujii H., Kato K., Horimoto T., Kyuwa S., Yoshikawa Y., Akashi H. Genomic and serological detection of bat coronavirus from bats in the Philippines. Arch. Virol. 2012;157:2349–2355. doi: 10.1007/s00705-012-1410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boheemen S., de Graaf M., Lauber C., Bestebroer T.M., Raj V.S., Zaki A.M., Osterhaus A.D., Haagmans B.L., Gorbalenya A.E., Snijder E.J., Fouchier R.A. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio. 2012;3 doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L. Human coronaviruses: what do they cause? Antivir. Ther. 2007;12:651–658. [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Berkhout B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol. Rev. 2006;30:760–773. doi: 10.1111/j.1574-6976.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykrishna D., Smith G.J., Zhang J.X., Peiris J.S., Chen H., Guan Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007;81:4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L., Keyaerts E., Lemey P., Maes P., Van Reeth K., Nauwynck H., Pensaert M., Van Ranst M. Evolutionary history of the closely related group 2 coronaviruses: porcine hemagglutinating encephalomyelitis virus, bovine coronavirus, and human coronavirus OC43. J. Virol. 2006;80:7270–7274. doi: 10.1128/JVI.02675-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L., Keyaerts E., Moes E., Thoelen I., Wollants E., Lemey P., Vandamme A.M., Van Ranst M. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005;79:1595–1604. doi: 10.1128/JVI.79.3.1595-1604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee S., Boongird K., Wanghongsa S., Ratanasetyuth N., Supavonwong P., Saengsen D., Gongal G.N., Hemachudha T. A longitudinal study of the prevalence of Nipah virus in Pteropus lylei bats in Thailand: evidence for seasonal preference in disease transmission. Vector Borne Zoonotic Dis. 2010;10:183–190. doi: 10.1089/vbz.2008.0105. [DOI] [PubMed] [Google Scholar]
- Wacharapluesadee, S., Sintunawa, C., Kaewpom, T., Khongnomnan, K., Olival, K.J., Epstein, J.H., 2013. Group C betacoronavirus in bat guano fertilizer, Thailand. Emerg. Infect. Dis. (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Watanabe S., Masangkay J.S., Nagata N., Morikawa S., Mizutani T., Fukushi S., Alviola P., Omatsu T., Ueda N., Iha K., Taniguchi S., Fujii H., Tsuda S., Endoh M., Kato K., Tohya Y., Kyuwa S., Yoshikawa Y., Akashi H. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 2010;16:1217–1223. doi: 10.3201/eid1608.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiology and molecular biology reviews: MMBR. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Chu C.M., Chan K.H., Tsoi H.W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.K., Poon L.L., Wong S.S., Guan Y., Peiris J.S., Yuen K.Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Huang Y., Yuen K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Lam C.S., Lau C.C., Tsang A.K., Lau J.H., Bai R., Teng J.L., Tsang C.C., Wang M., Zheng B.J., Chan K.H., Yuen K.Y. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Li K.S., Poon R.W., Wong B.H., Tsoi H.W., Yip B.C., Huang Y., Chan K.H., Yuen K.Y. Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Wang M., Lau S.K., Xu H., Poon R.W., Guo R., Wong B.H., Gao K., Tsoi H.W., Huang Y., Li K.S., Lam C.S., Chan K.H., Zheng B.J., Yuen K.Y. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007;81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Peng G., Wilken M., Geraghty R.J., Li F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012;287:8904–8911. doi: 10.1074/jbc.M111.325803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Ren X., Yang L., Hu Y., Yang J., He G., Zhang J., Dong J., Sun L., Du J., Liu L., Xue Y., Wang J., Yang F., Zhang S., Jin Q. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012;86:10999–11012. doi: 10.1128/JVI.01394-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R.H., He J.F., Evans M.R., Peng G.W., Field H.E., Yu D.W., Lee C.K., Luo H.M., Lin W.S., Lin P., Li L.H., Liang W.J., Lin J.Y., Schnur A. Epidemiologic clues to SARS origin in China. Emerg. Infect. Dis. 2004;10:1030–1037. doi: 10.3201/eid1006.030852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Wu Z., Ren X., Yang F., He G., Zhang J., Dong J., Sun L., Zhu Y., Du J., Zhang S., Jin Q. Novel SARS-like Betacoronaviruses in Bats, China, 2011. Emerg. Infect. Dis. 2013;19:989–991. doi: 10.3201/eid1906.121648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Hon C.C., Li Y., Wang D., Xu G., Zhang H., Zhou P., Poon L.L., Lam T.T., Leung F.C., Shi Z. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 2010;91:1058–1062. doi: 10.1099/vir.0.016378-0. [DOI] [PubMed] [Google Scholar]
- Yuen K.Y., Lau S.K., Woo P.C. Wild animal surveillance for coronavirus HKU1 and potential variants of other coronaviruses. Hong Kong Med. J. 2012;18(Suppl. 2):25–26. [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zeng Q., Langereis M.A., van Vliet A.L., Huizinga E.G., de Groot R.J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. USA. 2008;105:9065–9069. doi: 10.1073/pnas.0800502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.M., Herbst W., Kousoulas K.G., Storz J. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J. Med. Virol. 1994;44:152–161. doi: 10.1002/jmv.1890440207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkel F., Kurth A., Quan P.L., Briese T., Ellerbrok H., Pauli G., Leendertz F.H., Lipkin W.I., Ziebuhr J., Drosten C., Junglen S. An insect nidovirus emerging from a primary tropical rainforest. MBio. 2011;2:e00011–e00077. doi: 10.1128/mBio.00077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]