


Abstract
The database of Clusters of Orthologous Groups of proteins (COGs), which represents an attempt on a phylogenetic classification of the proteins encoded in complete genomes, currently consists of 2791 COGs including 45 350 proteins from 30 genomes of bacteria, archaea and the yeast Saccharomyces cerevisiae (http://www.ncbi.nlm.nih.gov/COG). In addition, a supplement to the COGs is available, in which proteins encoded in the genomes of two multicellular eukaryotes, the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster, and shared with bacteria and/or archaea were included. The new features added to the COG database include information pages with structural and functional details on each COG and literature references, improvements of the COGNITOR program that is used to fit new proteins into the COGs, and classification of genomes and COGs constructed by using principal component analysis.
INTRODUCTION
The database of Clusters of Orthologous Groups of proteins (COGs) has been incepted as a phylogenetic classification of proteins from complete genomes (1). Each COG includes proteins that are thought to be orthologous, i.e. connected through vertical evolutionary descent (2). Orthology may involve not only one-to-one, but also, in cases of lineage-specific gene duplications, one-to-many and many-to-many relationships (hence Orthologous Groups of proteins). The purpose of the COGs database is to serve as a platform for functional annotation of newly sequenced genomes and for studies on genome evolution. To facilitate functional studies, the COGs have been classified into 17 broad functional categories, including a class for which only a general functional prediction, usually that of biochemical activity, was feasible and a class of uncharacterized COGs. Additionally, some of the COGs with known functions are organized to represent specific cellular systems and biochemical pathways. The database is accompanied by the COGNITOR program, which assigns new proteins, typically from newly sequenced genomes, to pre-existing COGs. Here we describe the new developments in the COGs database in the year 2000, which included both the quantitative update through addition of new genomes and development of new functionalities associated with the database.
THE CURRENT STATUS OF THE COGS—NEW GENOMES
Since the second release of the COG database in January 2000 (3), nine new genomes have been added to the database using the COGNITOR program with subsequent manual validation to identify new members of pre-existing COGs and previously described procedures for the construction of new COGs. The additions included the first sequenced genome of a crenarchaeon (representative of the second major division of the archaea), Aeropyrum pernix; a fifth representative of the Euryarchaea, Pyrococcus abyssi; and seven bacterial genomes, including those from unusual organisms such as the extremely radioresistant Deinococcus radiodurans (Table 1). The previously described trend held with the new genomes in that 60–80% of the proteins from each of the prokaryotic genomes could be included in COGs (Table 1).
Table 1. Representation of genomes in the COGsa.
| Species |
Total no. of encoded proteins |
No. of proteins assigned to COGs |
Proteins in COGs (%) |
| Archaea | |||
| Archaeoglobus fulgidus | 2420 | 1817 | 75 |
| Methanococcus jannaschii | 1786 | 1301 | 74 |
| Methanobacterium thermoautotrophicum | 1873 | 1365 | 73 |
| P.abyssi | 1767 | 1430 | 81 |
| Pyrococcus horikoshiib | 2080 | 1353 | 66 |
| A.pernixb | 2722 | 1157 | 43 |
| Bacteria | |||
| Aquifex aeolicus | 1560 | 1312 | 84 |
| Bacillus subtilis | 4118 | 2767 | 67 |
| Borrelia burgdorferic | 1637 | 693 | 43 |
| Campylobacter jejuni | 1634 | 1282 | 78 |
| Chlamydia trachomatis | 895 | 630 | 71 |
| Chlamydia pneumoniae | 1053 | 646 | 62 |
| D.radiodurans | 3194 | 2133 | 67 |
| Escherichia coli | 4285 | 3308 | 77 |
| Haemophilus influenzae | 1695 | 1497 | 88 |
| Helicobacter pylori | 1578 | 1070 | 68 |
| Mycobacterium tuberculosis | 3924 | 2456 | 63 |
| Mycoplasma genitalium | 471 | 374 | 79 |
| Mycoplasma pneumoniae | 680 | 419 | 62 |
| Neisseria meningitidis | 2081 | 1446 | 70 |
| Pseudomonas aeruginosa | 5567 | 4166 | 75 |
| Rickettsia prowazekii | 836 | 673 | 81 |
| Synechocystis sp. | 3168 | 2048 | 65 |
| Thermotoga maritima | 1858 | 1497 | 81 |
| Treponema pallidum | 1036 | 705 | 68 |
| Vibrio cholerae | 3828 | 2715 | 71 |
| Ureaplasma urealyticum | 613 | 398 | 64 |
| Xylella fastidiosa | 2766 | 1481 | 54 |
| Eukaryotes | |||
| S.cerevisiae | 5964 | 2158 | 36 |
| Total | 68571 | 45350 | 66 |
aNewly added genomes are underlined.
bThe low fraction of proteins assigned to COGs is probably due to over-prediction of protein-coding genes in the original genome annotation (see text and Table 2)
cThe low fraction of proteins assigned to COGs is due to the fact that part of the genome consists of multiple plasmids that code for poorly conserved proteins
The genome of the crenarchaeon A.pernix (4), which was of particular interest because this major evolutionary lineage had not been previously represented among completely sequenced genomes, was investigated in detail as a benchmark for annotation of newly sequenced genomes using the COG system (5). The COG analysis resulted in an ∼50% increase in confident functional prediction for A.pernix genes compared to the original annotations. On the other hand, a significant fraction of open reading frames (ORFs), originally annotated as genes, did not show detectable similarity to any proteins in current databases, but overlapped with proteins included in the COGs, strongly suggesting that these ORFs were not real genes (Table 2). Thus the analysis of the genome of an organism that had no close relatives among other organisms with sequenced genomes appears to corroborate the effectiveness of the COG system as a genome annotation tool.
Table 2. Analysis of the predicted A.pernix proteins using the COG system.
| Originally predicted | Proteins assigned to COGs | Original ORFs overlapping | Predicted proteins after | ||
| |
proteins |
Predicted function |
Function unknown |
with COG members |
COG analysis |
| Number | 2722 | 833 | 315 | 849 | 1843 |
| %original gene set | 100 | 31 | 12 | 32 | 68 |
| % gene set after COG analysis | 146 | 45 | 17 | NA | 100 |
NA, not applicable.
Given the accumulation of multiple, complete genome sequences, we were interested in the growth dynamics of the COG set with the increased number of included genomes. The growth curve was constructed by imitating the COG formation for each of the 106 random orders of genome inclusion (Fig. 1). For each number of species, the maximum, the minimum and the average number of COGs was determined. The minimal and the maximal curves define the area containing all possible growth curves (Fig. 1). The average curve approximates the expected dynamics of the COG growth. Given that the number of completely sequenced genomes is still relatively small and that some of them are closely related, it remains uncertain whether or not the number of COGs is starting to approach saturation, and if it is, what is the asymptotic value.
Figure 1.
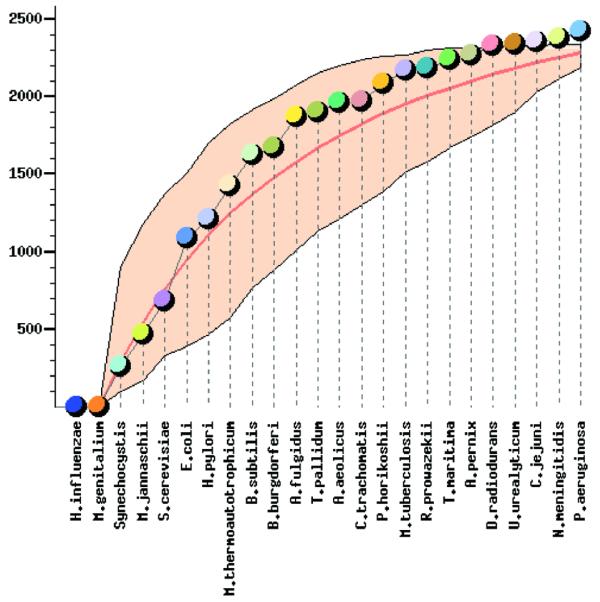
Growth dynamics of the COG set with the increase of number of included genomes. The circles show the sequence of genome inclusion according to the actual order of sequencing, and the smooth line shows the mean of 106 random permutations of the genome order. The colored area indicates the range between the maximal and minimal value for each point (number of genomes) in 106 random permutations.
ADDING PROTEINS FROM MULTICELLULAR EUKARYOTES TO PROKARYOTIC COGs
The current COG collection includes multiple bacterial and archaeal genomes and only one eukaryotic species, the yeast Saccharomyces cerevisiae. Incorporating the larger genomes of multicellular eukaryotes into the COG system is a challenging task due to the preponderance of multidomain proteins in these organisms. As a first step toward this goal, we sought to identify eukaryotic proteins that fit into already existing COGs, in other words, those eukaryotic proteins that have orthologs in at least two prokaryotic species. To this end, 19 895 protein sequences from the (nearly) complete genome of the nematode Caenorhabditis elegans (6) and 14 100 sequences from the genome of the fruit fly Drosophila melanogaster (7) were analyzed using the COGNITOR program, which assigns proteins to COGs on the basis of multiple genome-specific best hits and splits multidomain protein into individual domains if these show affinity with different COGs. After manual validation of the results, 20% of the D.melanogaster proteins and 14% of the C.elegans proteins were assigned to COGs; a significant number of proteins from each of the multicellular eukaryotes were included in COGs of each functional category, with the notable exception of ‘Cell division and chromosome partitioning’ and ‘Cell motility and secretion’, which consist primarily of prokaryote-specific proteins (Table 3). The COG analysis of the worm and fly proteins yielded numerous functional predictions, which have not been described previously (I.V.Garkavtsev and E.V.Koonin, unpublished observations). Eukaryotic proteins that have orthologs in prokaryotes belong to two major categories: (i) ancient proteins inherited from the last common ancestor of all extant life forms or at least the common ancestor of archaea and eukaryotes; (ii) proteins encoded by genes that have been horizontally transferred from organelles to the eukaryotic nucleus or otherwise acquired by eukaryotes from bacteria (8). Analysis of the phylogenetic patterns in the COGs may help distinguish between these two categories.
Table 3. Eukaryotic proteins in the COGs.
| Functional category | Eukaryotic proteins assigned to COGs | ||
| |
S.cerevisiae |
C.elegans |
D.melanogaster |
| Translation | 276 | 221 | 270 |
| Transcription | 107 | 134 | 167 |
| Replication and repair | 165 | 186 | 159 |
| Post-translational modification, chaperone functions | 167 | 260 | 273 |
| Cell division and chromosome partitioning | 23 | 22 | 19 |
| Cell motility and secretion | 10 | 17 | 14 |
| Cell envelope biogenesis, outer membrane | 29 | 62 | 47 |
| Inorganic ion transport | 85 | 199 | 132 |
| Signal transduction | |||
| Energy production and conversion | 116 | 138 | 183 |
| Carbohydrate transport and metabolism | 176 | 295 | 300 |
| Amino acid transport and metabolism | 180 | 193 | 222 |
| Nucleotide transport and metabolism | 85 | 88 | 99 |
| Coenzyme metabolism | 86 | 63 | 69 |
| Lipid metabolism | 52 | 237 | 169 |
| General function prediction only | 344 | 673 | 635 |
| Function unknown | 53 | 60 | 84 |
| Total | 1954 | 2848 | 2842 |
After three distant eukaryotic genomes were included in the prokaryotic COGs, it was of interest to analyze their co-occurrence. As expected, the majority of COGs with eukaryotic members include all three genomes; at the same time, a considerable number of COGs include all possible pairs of eukaryotic genomes and each of the individual species (Table 4). These observations, which will be analyzed in detail elsewhere, support the major role of lineage-specific gene loss and horizontal gene transfer in eukaryotic evolution.
Table 4. Co-occurrence of the eukaryotic genomes in the COGs.
| Numbers of COGs | Eukaryotic species | ||
| 578 | C.elegans | D.melanogaster | S.cerevisiae |
| 99 | C.elegans | D.melanogaster | |
| 38 | C.elegans | S.cerevisiae | |
| 46 | C.elegans | ||
| 77 | D.melanogaster | S cerevisiae | |
| 44 | D.melanogaster | ||
| 166 | S.cerevisiae |
DETECTING MISSED GENES
One of the features associated with the COG database is the analysis of phylogenetic patterns, i.e. the patterns of species that are represented or not represented in each of the COGs. Unexpected phylogenetic patterns, for example, those that contain all but one bacterial species or those that include only one of a pair of closely related species, may be due to omission of genes in genome annotations submitted to GenBank or to unusual evolutionary phenomena such as non-orthologous displacement of a nearly ubiquitous gene. Before considering the second hypothesis, the first one should be tested, and we undertook a systematic analysis of COGs with unexpected phylogenetic patterns in search of missing members (9). The nucleotide sequence of the genome in question was searched using the TNBLASTN program (10) and the sequences of members of the respective COGs as queries. As a result, missing genes coding for members of 48 COGs were identified (Table 5); most of the predicted new proteins are small, which explains why they have escaped the original genome annotations. Thus the COG system is instrumental in improving genome annotation not only with respect to functional predictions, but also for gene identification per se.
Table 5. Detection of missed proteins using phylogenetic pattern analysis.
| Species | Number of previously undetected | COGs including new proteins |
| proteins assigned to COGs | ||
| A.fulgidus | 3 | 1143, 1255, 1698 |
| M.jannaschii | 4 | 0286, 0827, 1908, 1996 |
| M.thermoautotrophicum | 1 | 2888 |
| P.abyssi | 1 | 2888 |
| P.horikoshii | 15 | 1383, 1761, 1919, 1998, 2004 |
| 2051, 2075, 2092, 2093, 2097 | ||
| 2167, 2212, 2260, 2443, 2888 | ||
| A.pernix | 19 | 0640, 1522, 1605, 1694, 1848 |
| 1858, 2002, 2118, 2260, 2443 | ||
| 2888 | ||
| A.aeolicus | 6 | 0254, 0255, 0690, 0858, 1828 |
| 2608 | ||
| B.subtilis | 2 | 1582, 1863 |
| C.trachomatis | 1 | 1314 |
| D.radiodurans | 2 | 1863, 2120 |
| H.influenzae | 1 | 1826 |
| H.pylori | 1 | 0690 |
| M.tuberculosis | 1 | 0458 |
| M.genitalium | 1 | 0828 |
| M.pneumoniae | 3 | 0816, 0828, 1546 |
| T.maritima | 2 | 0230, 1886 |
| T.pallidum | 1 | 0268 |
NEW FEATURES ASSOCIATED WITH THE COGS
Improvement of the COGNITOR program—statistical evaluation of the fit
The original COGNITOR program uses multiple genome-specific best hits (BeTs) as the only criterion for assigning new proteins to COGs. In the new version, we introduced an estimate of the probability that the query protein is assigned to the given COG by chance. Under the assumption of uniform distribution of hits to each genome in the COG database, the probability of one BeT into a particular COG is, simply, the fraction of proteins from the specified genome that belongs to the COG:
fij = nij/Ni
where nij is the number of proteins from species i in COG j and Ni is the total number of proteins in species i. Then, the probability of exactly two BeTs into COG j is given by:
p2j = 1/2Σfijfkj(1 – flj)
Similar expressions can be easily obtained for a different number of BeTs. For each COG, we can compute p2j and find the ‘average’ value of Fj that satisfies the equation:
C(2,m)Fj2 (1 – Fj)(m–2) = p2j
where m is the number of species in COG j. Using Fj simplifies the calculation of the probability when the specified number of BeTs is large.
COG-Info pages
In order to increase the utility of the COG system for genome annotation, a web page that contains additional structural and functional information on the COG as a whole and individual members is now associated with each COG. These hyperlinked Info pages include: systematic classification of the COG members under the current classification systems for enzyme or transporters (if applicable); indications which COG members (if any) have been characterized genetically and biochemically; information on the domain architecture of the proteins comprising the COG and the three-dimensional structure of the domains if known or predictable; a succinct summary of the common structural and functional features of the COG members and peculiarities of individual members; key references (Fig. 2). The COG-Info pages are currently at different stages of construction.
Figure 2.

An example of a COG-Info page.
Classification of genomes on the basis of co-occurrence in COGs using principal component analysis
The data on the co-occurrence of genomes in COGs was used as the input for classification by principal component analysis (PCA). Briefly, the presence or absence of a given species in each COG is converted into a 1/0 coordinate value in a multidimensional space where each dimension corresponds to a COG, which results in a geometric representation of all included species in the >2000-dimensional space. The PCA analysis is then used to choose the subspace of lower dimensionality for visual examination. The eigenvector decomposition yields the orthogonal courses in the space and the corresponding eigenvectors constitute the spread of the objects. The WWW interface provides tools for selection of the subspace, the species to view and the COGs to use for classification (Fig. 3A). Significantly different results were obtained when different functional categories of COGs were analyzed. Specifically, the combined categories of translation, transcription and replication showed a sharp separation between bacteria, archaea and eukaryotes, with representatives of each of these primary domains of life forming a tight cluster (Fig. 3B); the metabolic functions produced a more complex picture, with a separation of free-living and parasitic bacteria and grouping of yeast with the former (Fig. 3C).
Figure 3.

Classification of genome by co-occurrence in COGs using PCA. (A) All COGs. (B) Translation, transcription and replication (functional categories J, K and L). (C) Metabolism (functional categories C, E, F, G, H and I).
Integration of COGs with the Genome Division of Entrez
The COGs are now integrated with the Genomes division of the Entrez system. From the COG pages, the proteins are linked to the Entrez genome view (the ‘Genome’ button) and to the protein neighbor view (the Blink button). Conversely, the Genomes division of Entrez (11) incorporates COG information in several displays. The COG information including the breakdown by the functional categories is presented for each genome, for example: http://www.ncbi.nlm.nih.gov:80/cgi-bin/Entrez/coxik?gi=131. The main page for each genome includes a (usually) circular genome map, with radial lines corresponding to genes color-coded according to the functional categories adopted in the COG system. Additionally, for all proteins that belong to COGs, the protein view is linked to the respective COG.
THE COG WORLDWIDE WEB SITE
The COG database is accessible at http://www.ncbi.nlm.nih.gov/COG. The site includes the following main features: complete list of all COGs hyperlinked to individual COG pages; COGs organized by functional category; COGs organized by functional complexes and pathways; an interactive matrix of co-occurrence of genomes in COGs; a phylogenetic pattern search tool; a principal component classification tool; COGNITOR; a COG Help page. Each of the individual COG pages is hyperlinked to: (i) pictorial representations of BLAST search outputs for each member of the COG, which also include links to the respective GenBank and Entrez-Genomes entries, (ii) a multiple alignment of the COG members produced automatically by using the ClustalW program, (iii) a COG-Info page (reached by clicking on the COG number). The supplement to the COGs, which shows proteins from C.elegans and D.melanogaster assigned to each COG is accessible at http://www.ncbi.nlm.nih.gov/COG/euk. The COG data set is also available by anonymous ftp at ftp://ncbi.nlm.nih.gov/pub/COG.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to David Lipman for his critical contribution at the initial stage of the COG project and constant support and inspiration and to Vivek Anantharaman, L. Aravind, Kira Makarova, Igor Rogozin and Yuri Wolf for helpful suggestions.
References
- 1.Tatusov R.L., Koonin,E.V. and Lipman,D.J. (1997) A genomic perspective on protein families. Science, 278, 631–637. [DOI] [PubMed] [Google Scholar]
- 2.Fitch W.M. (1970) Distinguishing homologous from analogous proteins. Syst. Zool., 19, 99–106. [PubMed] [Google Scholar]
- 3.Tatusov R.L., Galperin,M.Y., Natale,D.A. and Koonin,E.V. (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res., 28, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawarabayasi Y., Hino,Y., Horikawa,H., Yamazaki,S., Haikawa,Y., Jin-no,K., Takahashi,M., Sekine,M., Baba,S., Ankai,A. et al. (1999) Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res., 6, 83–101. [DOI] [PubMed] [Google Scholar]
- 5.Natale D.A., Shankavaram,U.T., Galperin,M.Y., Wolf,Y.I., Aravind,L. and Koonin,E.V. (2000) Genome annotation using clusters of orthologous groups of proteins (COGs) – towards understanding the first genome of a Crenarchaeon. Genome Biol., 1, 0009.1–0009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. The C.elegans Sequencing Consortium. (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. The C.elegans Sequencing Consortium. Science, 282, 2012–2018. [DOI] [PubMed] [Google Scholar]
- 7.Adams M.D., Celniker,S.E., Holt,R.A., Evans,C.A., Gocayne,J.D., Amanatides,P.G., Scherer,S.E., Li,P.W., Hoskins,R.A., Galle,R.F. et al. (2000) The genome sequence of Drosophila melanogaster. Science, 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 8.Doolittle W.F. (1998) You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet., 14, 307–311. [DOI] [PubMed] [Google Scholar]
- 9.Natale D.A., Galperin,M.Y., Tatusov,R.L. and Koonin,E.V. (2000) Using the COG database to improve gene recognition in complete genomes. Genetica, 108, 9–17. [DOI] [PubMed] [Google Scholar]
- 10.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tatusova T.A., Karsch-Mizrachi,I. and Ostell,J.A. (1999) Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics, 15, 536–543. [DOI] [PubMed] [Google Scholar]