

Abstract
The steroid receptor coactivator (SRC)-1 isoform/estrogen receptor (ER)-β axis has an essential role in endometriosis progression. In this context, therefore, bufalin was employed as a ‘tool compound’ to evaluate inhibitors of SRC in alternative endometriosis treatment. Bufalin effectively suppressed the growth of primary human endometrial stroma cells isolated from endometriosis patients compared to women without endometriosis and immortalized human endometrial epithelial and stromal cells expressing the SRC-1 isoform compared to their parental cells in vitro. In vivo, compared to the vehicle, bufalin treatment significantly suppressed the growth of endometriotic lesions in mice with surgically induced endometriosis because bufalin disrupted the functional axis of SRC-1 isoform/ERβ by increasing SRC-1 isoform protein stability, hyperactivating the transcriptional activity of the SRC-1 isoform, and degrading the ERβ protein by proteasome 26S subunit, non-ATPase 2 in endometriotic lesions. Bufalin treatment elevated the apoptosis signaling in epithelial cells of endometriotic lesions. In stromal cells of endometriotic lesions, bufalin treatment increased the levels of pyroptosis markers (caspase 1 and the active form of interleukin 1β) and reduced proliferation. In addition, bufalin treatment increased the expression levels of endoplasmic reticulum (ER)-stress markers (PKR-like ER kinase, protein disulfide isomerase and binding immunoglobulin) in endometriotic lesions. Collectively, the bufalin-induced disruption of the SRC-1 isoform/ERβ axis might induce apoptosis, pyroptosis and ER-stress signaling in endometriotic lesions, causing the suppression of endometriosis. Therefore, future generations of SRC-modulators could be employed as an alternative medical approach for endometriosis treatment.
Keywords: Endometriosis, Steroid Receptor Coactivator 1 Isoform, Estrogen Receptor β, Bufalin, Apoptosis, Pyroptosis
Introduction
As an estrogen-dependent pro-inflammatory disease, endometriosis is defined as the colonization and growth of endometrial tissues at anatomic sites outside of the uterine cavity, primarily in the pelvic peritoneum and ovaries (Bulun 2009). Up to 10% of reproductive-aged women in the United States chronically suffer from symptoms of endometriosis, which include pelvic pain, infertility, menstrual cycle abnormalities and increased risk of certain cancers, such as ovarian, breast and skin cancers (Brilhante, et al. 2017; Farland, et al. 2017; Farland, et al. 2016; Poole, et al. 2017; Vercellini, et al. 2014).
Due to the severe chronic morbidity associated with this gynecological disorder, a number of past studies have attempted to identify the distinguishing molecular features of the endometriotic lesion with a view to developing more effective prognostic, diagnostic, and/or treatment strategies in the clinical management of this debilitating disease (Bedaiwy, et al. 2017). Despite such efforts, however, many of the current clinical treatments are not adequately effective at treating this disease and produce unacceptable side-effects. For example, studies have shown that levels of prostaglandin E2 (PGE2), cyclooxygenase-2 (COX-2) and various cytokines are highly elevated in endometriotic tissue relative to the normal endometrium, supporting a heightened pro-inflammatory response as a major component of this disease (Hirata, et al. 2011; Sacco, et al. 2012). Therefore, selective COX-2 inhibitors are used as the conventional treatment for this disorder (Ebert, et al. 2005). However, COX-2 selective inhibitors have gastrointestinal side-effects, even though their side effects are much less severe than older non-steroidal anti-inflammatory drugs (Ebert et al. 2005).
Similarly, it has been well established that increased concentrations of 17β-estradiol (E2) in endometriotic tissues arise from locally elevated levels of aromatase along with reduced activity of 17β-hydroxysteroid dehydrogenase-2 (Bulun, et al. 2010; Lamp, et al. 2010). Therefore, along with the anti-inflammatory treatments described above, current endometriosis treatments also involve suppressing E2 levels through the use of gonadotropin-releasing hormone agonists, oral contraceptives, synthetic progestins and/or aromatase inhibitors (Goenka, et al. 2017). However, many clinical reports have revealed that these systemic estrogen deficiency therapies cause infertility and confer harmful side effects in other estrogen target tissues, such as bone and brain.
Because of the unacceptable deficiencies cited above, there is clearly an urgent need to identify new molecular mechanisms that critically underpin the initiation and progression of endometriosis to develop more effective therapeutics that lack the side effects of current treatments. Interestingly, our prior study revealed that a steroid receptor coactivator (SRC)-1 isoform that is proteolytically cleaved from the full-length SRC-1 by matrix metalloproteinase-9 and estrogen receptor (ER) β are specifically elevated in endometriotic tissues compared to the normal endometrium, and this SRC-1 isoform/ERβ axis has an essential role in endometriosis progression because this axis prevents TNFα-mediated apoptosis and enhances inflammasome-mediated inflammatory signaling in endometriotic lesions for their survival (Han, et al. 2012; Han, et al. 2015). Collectively, these recent findings led us to hypothesize that the SRC-1 isoform/ERβ axis should be a new molecular therapeutic target for an alternative endometriosis treatment to enhance the specificity of endometriosis treatment and reduce the side effects of previous endometriosis treatments. Our previous studies defined small molecular inhibitors (SMIs), such as bufalin and gossypol, that diminished the activities and protein stabilities of SRCs and suppressed the growth of various cancer cells (Wang, et al. 2011; Wang, et al. 2014). These observations led us to investigate whether SMIs against SRCs could be employed to suppress endometriosis progression due to the crucial role of the SRC-1 isoform in endometriosis progression. Therefore, we here show that bufalin, one of the SMIs against SRCs, represents a new class of drugs that could be used to combat endometriosis based on its antagonistic role against the SRC-1 isoform/ERβ axis in endometriotic lesions.
Materials and Methods
Mice
Mice were housed in a pathogen-free animal facility under a standard 12-h light/12-h dark cycle and were fed standard rodent chow and water. All animal experimentation was conducted in accordance with accepted standards of humane animal care. All animal care was controlled by the ethical regulations approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine. Five-week-old normal (C57BL/6J) mice were purchased from Jackson Laboratory.
Immortalized human endometrial cells
Primary human endometrial stromal cells isolated from women with/without endometriosis (Han et al. 2012), immortalized human endometrial stromal cells (IHESCs) (Krikun, et al. 2004), EMosis-CC/TERT1 (immortalized human endometriotic epithelial cells; IHEECs) (Bono, et al. 2012) and HeLa cells were confirmed by Short Tandem Repeat profiling; these cells were not contaminated with mycoplasma.
Generation of a lentivirus expressing the SRC-1 isoform
The open reading frame of the SRC-1 isoform gene was cloned into a pCDH-pCMV vector using BamH1 and XhoI restriction enzymes. Next, 293T cells on a 100-cm tissue culture dish were transfected with the pCDH-pCNV-SRC-1 isoform and Lenti-X packaging single shot (Clontech Laboratories, Inc., Mountain View, CA). The virus-containing medium was collected at 48 hours after transfection. The lentivirus titer was determined by Lenti-X GoStix (Clontech Laboratories, Inc., Mountain View, CA).
Generation of IHESCs and IHEECs that expressed the SRC-1 isoform
IHEECs and IHESCs were cultured in a 10-cm dish. When the cell confluency reached 70%, 6 ml of new medium containing 64 μg of Polybrene was added, and then 2 ml of media containing lentivirus (MOI of approximately 2) was added. At 2 days after transduction, 2 μg/ml of puromycin was added to the media. The puromycin-resistant cells were selected, and then the expression of the SRC-1 isoform in these cells was determined by Western blot analyses with SRC-1 antibody.
MTS cell growth assay
Primary human endometrial stromal cells isolated from women with/without endometriosis, IHEECs, IHEECs:SRC-1 ISO, IHESCs and IHESCs:SRC-1 ISO were inoculated into the wells of 96-well plates (1×104 cells/well). The next day, each cell line was treated with serially diluted bufalin (0- 800 nM) and vehicle as the control. After 2 days, 10 μL of MTS reagent was added to each well. MTS-treated plates were incubated for 2 more hours. After that, the optical density of color in each well was measured at 490 nm in a microtiter plate reader.
Surgically induced endometriosis
Endometriosis in mice was surgically induced under aseptic conditions under anesthesia using a modified method as described previously (Cummings and Metcalf 1995). Briefly, C57BL/6 mice were subjected to ovariectomy at six weeks old. After one week, the ovariectomized mice were implanted with a sterile, 60-day release pellet containing 0.36 mg of 17-β estradiol (Innovative Research of America, Sarasota, FL). Two days later, one uterine horn from each mouse was isolated under anesthesia. In a Petri dish containing warmed DMEM/F-12 supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin, the uterine horns were longitudinally cut with a pair of scissors. Next, using a 2-mm dermal biopsy punch, one endometrial fragment was isolated and subsequently sutured to the mesenteric membrane attached to the intestine in the same mouse through a midline incision (7-0 braided polypropylene suture). In the case of sham-treated control mice, a suture was performed without endometrial tissue fragments. The abdominal incision was then closed with a 5-0 braided polypropylene suture in a continuous fashion. On day 21 after endometriosis challenge, the mice were sacrificed, and the endometriotic lesions and eutopic endometria were carefully isolated from the surrounding tissue. Using the formula volume (mm3) = 0.52 × width × length × height, the volumes of the endometriotic lesions were calculated.
Bufalin treatment of endometriosis-induced mice
Endometriosis was surgically induced as described above. Based on a previous study, we injected mice with 1 mg/kg of bufalin (Zhang, et al. 2014). After endometriotic lesions were established (one week after endometriosis induction), the mice were randomly divided into two groups and then subcutaneously administered vehicle (as the control) or 1 mg/kg of bufalin daily for 21 days.
Bufalin treatment of wild-type mice
Female C57BL/6J (6 weeks old) were treated with 1 mg/kg of bufalin and vehicle as the control every day for 21 days. Two weeks before harvesting the uteri, mouse estrous cycles were determined using vaginal cytology (McLean, et al. 2012). At the estrus cycle after 21-day drug treatment, uteri were isolated from mice treated with bufalin and vehicle.
Fertility assay following bufalin treatment
C57BL/6J female mice (8 weeks of age) were treated daily with vehicle and bufalin (1.0 mg/kg) for 21 days (n=3/group). Afterwards, each female mouse was paired with a wild-type male of proven fertility (1:1). The fertility was assessed by monitoring the litter size over a two-month period.
Western blot analyses
Endometriotic tissues, human endometrial cells and transfected HeLa cells were washed with PBS solution and homogenized in a buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2.5 mM EDTA, and 0.5% Nonidet P-40 (vol/vol). Cellular debris was removed by centrifugation at 14,000 rpm for 15 min at 4 °C. The protein concentration was determined by Bradford’s method using bovine serum albumin as the standard. Samples containing 10 μg total proteins were subjected to 10% SDS-polyacrylamide gel electrophoresis. The separated proteins were then transferred onto a polyvinylidene difluoride membrane. Membranes were blocked overnight with 5% skim milk (wt/vol) in phosphate-buffered saline with 0.1% Tween 20 (vol/vol). Primary antibodies against the following proteins were used: SRC-1 (ab10308; Abcam), tubulin (SC-9104; Santa Cruz Biotechnologies), ERα (SC-542; Santa Cruz Biotechnologies), and chicken anti-ERβ antibody 503 (Saji, et al. 2000). Membrane-containing proteins were incubated with secondary horseradish peroxidase-tagged antibodies (Sigma), and the signals were visualized using an enhanced luminol-based substrate for horseradish peroxidase.
Immunohistochemistry
Immunostaining was performed with 10% neutral-buffered, formalin-fixed and paraffin-embedded sections of mouse tissue, as previously described (Han, et al. 2005). For immunostaining, sections were dewaxed, rehydrated, and boiled for 10 min in 10 mM citrate buffer, pH 6.0. To reduce nonspecific binding of antibodies, sections were washed in PBS again and preincubated with 5% BSA in PBS for 1 h at room temperature. Antibodies against PSMD2 (A303-854A-T; Bethyl Laboratories), UBA7 (ab133479, Abcam), Ki-67 (ab16667; Abcam), cleaved caspase 3 (9664; Cell Signal), caspase 1 (2225; Cell Signal), active form of Interleukin (IL)-1β (521875; Cell Signaling), PERK (5683; Cell Signal), PDI (3501; Cell signal), and BiP (3177; Cell Signal) were used. The specific antigens were visualized with the DAB substrate kit. The immunostaining intensity was quantified using the ImageJ program, which was developed by the National Institutes of Health.
TUNEL Assay
The TUNEL assay was conducted with a TACS.XL DAB In Situ Apoptosis Detection Kit and its protocol (Trevigen, Inc., Gaithersburg, MD).
Transfection and luciferase reporter gene assay
Transfections with plasmids were performed using Lipofectamine 2000 reagent (Invitrogen, Carlabad, CA) according to the manufacturer’s instructions. HeLa cells were transfected with the indicated expression plasmids. For the determination of ERβ transcriptional activity, estradiol (10−8 M) was added to cells 24 h following transfection and incubated for another 24 h. The cells were harvested, and the luciferase activity was determined and normalized against the total input protein.
Statistical analyses
Statistical analyses were performed by using Windows GraphPad Prism 5 (GraphPad Software, Inc.). The data are expressed as the mean ± SEM. Significance was assessed using an independent two-tailed Student’s t test. A P value of less than 0.05 was considered statistically significant. N.S., non-specific.
Results
Bufalin effectively inhibited the growth of primary human endometrial stromal cells isolated from endometriosis patients and immortalized human endometrial cells expressing the SRC-1 isoform
Compared to levels in the normal endometrium, levels of the SRC-1 isoform are highly elevated in endometriotic tissues, and this isoform prevents TNFα-induced apoptosis signaling in endometriotic lesions for their survival (Han et al. 2012). Therefore, targeting the SRC-1 isoform should reactivate TNFα-induced apoptosis in endometriotic lesions, killing them. To inhibit SRC activity, we previously identified SMIs that inhibited the function of SRCs. Bufalin, one of the SRC-SMIs, inhibited the intrinsic transcriptional activity of SRC-1 and -3 and degraded their proteins in various cancer cells to suppress their growth (Wang et al. 2014). These observations led us to examine whether bufalin might suppress endometriosis progression by inhibiting the function of the SRC-1 isoform in endometriotic tissues.
To determine whether endometriotic tissues are more sensitive to bufalin compared to normal endometrium, we determined the growth pattern of primary human endometrial stromal cells isolated from women with/without endometriosis upon bufalin treatment (Han et al. 2012). Bufalin effectively inhibited the growth of primary human endometrial stromal cells from endometriosis patients compared to those isolated from women without endometriosis (Fig. 1A). Therefore, endometriotic cells were more sensitive to bufalin treatment than normal endometrial cells.
Figure 1. Bufalin inhibited the growth of primary human endometrial stromal cells isolated from endometriosis patients and immortalized human endometrial cells expressing the SRC-1 isoform.
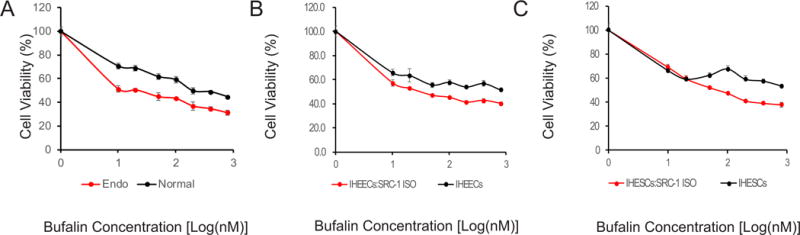
(A) The growth patterns of primary human endometrial stromal cells isolated from women with (Endo) and without (Normal) endometriosis were determined at different concentrations of bufalin (0, 10, 20, 50, 100, 200, 400 and 800 nM) for 48 h of treatment, by using the MTS cell growth assay. (B) The growth patterns of IHEECs expressing the SRC-1 isoform (IHEECs:SRC-1 ISO) and their parental cells were determined at different concentrations of bufalin for 48 h, by using the MTS cell growth assay. (C) The growth patterns of IHESCs expressing the SRC-1 isoform (IHESCs:SRC-1 ISO) and their parental cells were determined at different concentrations of bufalin for 48 h, by using the MTS cell growth assay. Data are presented as the means ± SEM.
In addition to primary human endometrial cells, we employed IHESCs and IHEECs as human endometrial epithelial and stromal cell lines, respectively, because IHESCs are karyotypically, morphologically, and phenotypically similar to the primary parent cells (Krikun et al. 2004) and because IHEECs also retain the normal functions and characteristics of the primary cells (Kyo, et al. 2003). To determine the role of the SRC-1 isoform in bufalin-mediated suppression of endometriotic cells, we generated recombinant IHEECs and IHESCs that stably expressed the SRC-1 isoform (IHEECs:SRC-1 ISO and IHESC:SRC-1 ISO) using a lentivirus containing the SRC-1 isoform gene expression unit. The growth of IHESCs:SRC-1 ISO and IHEECS:SRC-1 ISO was significantly reduced by bufalin treatment compared to their parental cells (Figs. 1B and 1C). Therefore, the overexpression of the SRC-1 isoform in human endometrial epithelial and stromal cells increased the sensitivity against bufalin.
Bufalin suppressed the growth of endometriotic lesions in mice with endometriosis in vivo
We next examined whether bufalin suppresses the growth of endometriotic lesions in mice with surgically induced endometriosis in vivo because of the essential role of the SRC-1 isoform in endometriosis progression. To address this issue, endometriosis was surgically induced in C57BL/6J mice using an autotranslation method. After the establishment of endometriotic lesions in mice (at one week after endometriosis induction), mice with endometriosis were randomly divided two groups and then injected with bufalin (1.0 mg/kg, daily, n=4/group) or with vehicle for the control (n=4/group). To determine the effect of bufalin in endometriosis progression, we isolated endometriotic lesions from each drug-treated mouse with endometriosis and then determined the volume of the endometriotic lesions. Compared to the vehicle, bufalin treatment reduced the volume of the endometriotic lesions by 14-fold (p=0.009) (Fig. 2A). Therefore, bufalin treatment significantly suppressed the growth of endometriotic lesions in mice with endometriosis in vivo.
Figure 2. Bufalin suppressed the growth of endometriotic lesions in mice with endometriosis.
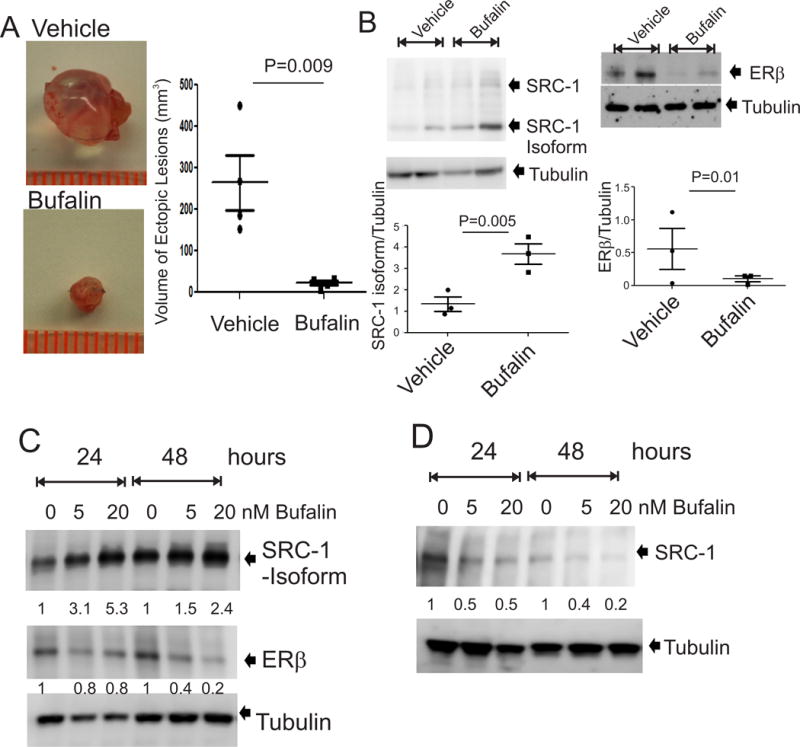
(A) Endometriotic lesions were isolated from mice with surgically induced endometriosis treated with 1.0 mg/kg bufalin and the vehicle as the control (n=4 mice for each group). The volume of each endometriotic lesion was determined by the formula volume (mm3) = 0.52 × width × length × height. (B) Protein levels of the SRC-1 isoform, ERβ and tubulin were determined in endometriotic lesions treated with bufalin (1 mg/kg) or vehicle by Western blot analysis (n=3 mice for each group). (C) IHESCs:SRC-1 ISO cells were treated with 5 and 20 nM bufalin for 24 and 48 h, and then protein levels of the SRC-1 isoform, ERβ and tubulin were determined by Western blot analysis. (D) IHESCs were treated with 5 and 20 nM bufalin for 24 and 48 h, and then protein levels of the full-length SRC-1 isoform and tubulin were determined by Western blot analysis. Data are presented as the means ± SEM and P value (Student’s t-test).
Since the SRC-1 isoform/ERβ axis plays a crucial role in endometriosis progression, we next examined whether bufalin treatment disrupts this SRC-1 isoform/ERβ functional axis in endometriotic lesions to suppress their growth. Western blot analysis revealed that compared to the vehicle, bufalin treatment increased SRC-1 isoform protein levels by 2.7-fold (p=0.005) in endometriotic lesions (Fig. 2B). To validate this bufalin-induced elevation of SRC-1 isoform levels in endometriotic lesions, IHESCs:SRC-1 ISO cells were treated with bufalin (0, 5 and 20 nM) for 24 or 48 hours. We found that 20 nM bufalin treatment, compared with the vehicle, elevated the SRC-1 isoform protein levels by 2.4-fold in IHESCs:SRC-1 ISO with 48-hour treatment (Fig. 2C). To define the effect of bufalin on full-length SRC-1, endogenous SRC-1 levels in IHESCs treated with bufalin were determined by Western blot analyses. In contrast to the results for the SRC-1 isoform, 20 nM bufalin treatment, compared with the vehicle, reduced the levels of full-length SRC-1 by 5.0-fold in IHESCs with 48-hour treatment (Fig. 2D). The bufalin-induced degradation of full-length SRC-1 in various cancer cells was also reported in our previous study (Wang et al. 2014). Therefore, bufalin increased SRC-1 isoform protein levels but reduced the full-length SRC-1 protein levels in endometriotic tissues.
In addition to the SRC-1 isoform, ERβ also has an essential role in endometriosis progression. Therefore, we determined whether bufalin also affects the ERβ axis in endometriotic lesions. In contrast to the results for the SRC-1 isoform, however, bufalin treatment, compared to the vehicle, reduced ERβ protein levels by 7.2-fold (p=0.01) in endometriotic lesions (Fig. 2B). In addition to those of endometriotic lesions, ERβ levels in IHESCs:SRC-1 ISO were reduced by 5.0-fold by 20 nM bufalin at 48-hour treatment compared with the levels after vehicle treatment (Fig. 2C). Therefore, bufalin treatment disrupted the SRC-1 isoform/ERβ axis by increasing SRC-1 isoform protein levels, decreasing ERβ protein levels in endometriotic lesions, and then suppressing endometriosis progression.
Bufalin increased the intrinsic transcriptional activity of the SRC-1 isoform but not the ERβ activity
We asked whether bufalin also affects the intrinsic transcriptional activity of the SRC-1 isoform in addition to protein levels because bufalin inhibits the intrinsic transcriptional activity of full-length SRC-1 (Wang et al. 2014). To address this issue, we generated a mammalian expression vector for the chimeric SRC-1 isoform protein fused to the Gal4 DNA-binding domain (pBID-SRC-1 isoform) and then transiently transfected it into HeLa cells along with a pG5 luciferase reporter containing Gal4 DNA-binding elements. Compared to the empty expression vector control, the SRC-1 isoform had an intrinsic transcriptional activity, and its transcriptional activity was elevated by 5 nM bufalin treatment (3-fold, P=0.005) compared to the vehicle (Fig. 3A). Collectively, bufalin hyperactivated the SRC-1 isoform function in endometriotic lesions by stabilizing the SRC-1 isoform protein, stimulating its transcriptional activity.
Figure 3. Bufalin stimulated the intrinsic transcriptional activity of the SRC-1 isoform.
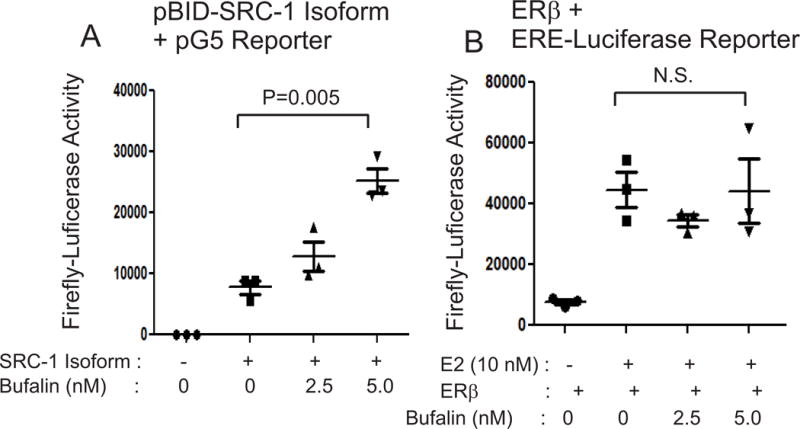
(A) HeLa cells were transfected with pBID-SRC-1 isoform and pG5 reporter vectors and then treated with 0, 2.5 or 5.0 nM bufalin for 48 h. The luciferase activity in HeLa cells treated with different doses of bufalin was determined to define the intrinsic transcriptional activity of the SRC-1 isoform upon bufalin treatment. (B) HeLa cells were transfected with an ERβ expression vector and ERE-luciferase reporter. To stimulate ERβ activity, HeLa cells were treated with estradiol (10 nM) plus 0, 2.5 or 5.0 nM bufalin treatment for 48 h. The luciferase activity in HeLa cells treated with different doses of bufalin was determined to define the intrinsic transcriptional activity of the ERβ upon bufalin treatment. Data are presented as the means ± SEM and P value (Student’s t-test).
We next determined the effect of bufalin on the intrinsic transcriptional activity of ERβ using an ERβ/Estrogen Response Element luciferase assay system in HeLa cells. Compared with the vehicle, estrogen treatment significantly enhanced the intrinsic transcriptional activity of ERβ (Fig. 3B). However, compared to the vehicle, bufalin treatment did not affect the E2-induced transcriptional activity of ERβ (Fig. 3B). Therefore, bufalin treatment decreased the protein levels of ERβ but did not inhibit its transcriptional activity in endometriotic lesions.
Bufalin degraded ERβ protein via the proteasome 26S subunit, non-ATPase 2 (PSMD2) in endometriotic tissues
How does bufalin degrade ERβ in endometriotic lesions? To determine the molecular function of ERβ in endometriosis progression, we isolated the ERβ-containing complex from endometriotic lesions using immunoprecipitation and then identified all protein components co-precipitated with ERβ (Han et al. 2015). Interestingly, these data revealed that several proteasome components, such as proteasome 26S Subunit, non-ATPase 2 (PSMD2) and ubiquitin-like modifier activating enzyme 7 (UBA7), were specifically co-precipitated with ERβ from endometriotic lesions. This observation led us to investigate the potential roles of PSMD2 and UBA7 in bufalin-induced ERβ protein degradation in endometriotic tissues. To validate this hypothesis, we measured the protein levels of PSMD2 and UBA7 in endometriotic lesions treated with bufalin versus vehicle using immunohistochemistry (IHC). Bufalin treatment increased the levels of PSMD2 and UBA7 in both epithelial and stromal cells from endometriotic lesions compared to vehicle-treated endometriotic lesions (Figs. 4A and 4B).
Figure 4. Bufalin degraded ERβ in endometriotic lesions via PSMD2.
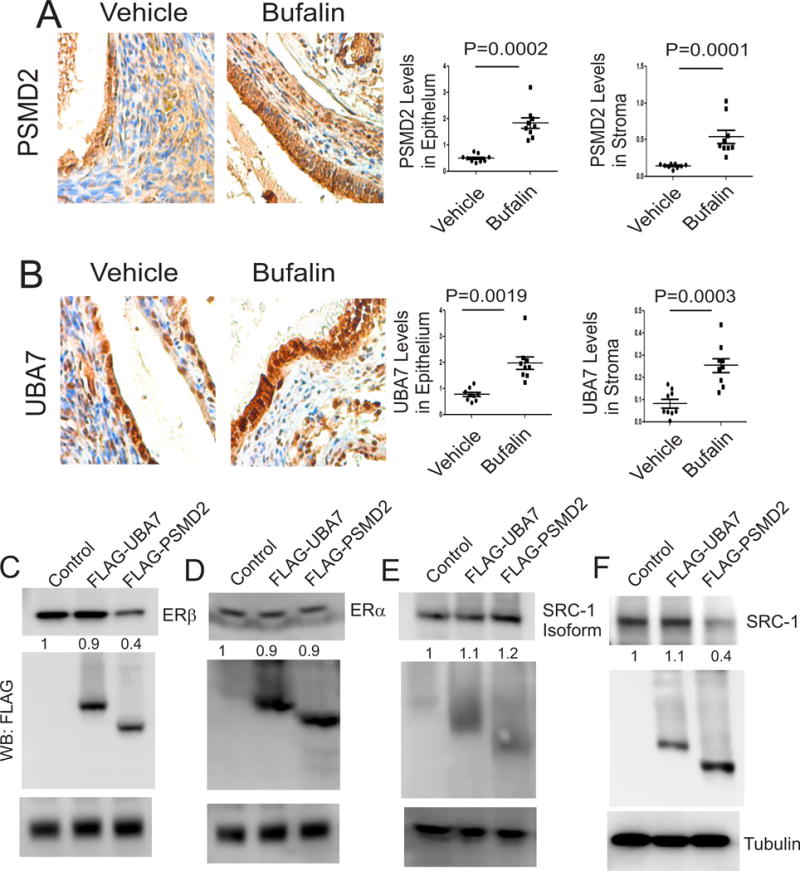
(A–B) Levels of PSMD2 (A) and UBA7 (B) proteins were determined in endometriotic lesions isolated from mice with endometriosis treated with bufalin (1 mg/kg) or vehicle by IHC. The expression levels of these proteins in epithelial cells and stromal cells of endometriotic lesions were quantified using ImageJ (n=3 mice for each group, three independent IHC assays from each mouse). (C) To determine ERβ degradation by UBA7 or PSMD2, HeLa cells were transfected with the ERβ expression vector with the control, UBA7 and PSMD2 expression vectors. (D) To define UBA7 or PSMD2-mediated ERα degradation, HeLa cells were transfected with the ERα expression vector with the control, UBA7 and PSMD2 expression vectors. (E) To determine whether UBA7 or PSMD2 degrades the SRC-1 isoform, HeLa cells were transfected with the SRC-1 isoform expression vector with the control, UBA7 and PSMD2 expression vectors. (F) To determine whether UBA7 or PSMD2 degrades full-length SRC-1, HeLa cells were transfected with the full-length SRC-1 expression vector with the control, UBA7 and PSMD2 expression vectors. At 48th h after transfection, protein levels of ERβ, ERα, full-length SRC-1, SRC-1 isoform and tubulin levels were determined by Western blot analyses with their antibodies. The protein levels of UBA7 and PSMD2 were determined by Western blot analyses with FLAG antibody because the proteins had a FLAG tag in the N-terminal region. Data are presented as the means ± SEM and P value (Student’s t-test).
We next examined whether the increased quantities of PSMD2 and UBA7 degraded ERβ proteins in endometriotic lesions. To address this issue, HeLa cells were cotransfected with expression vectors containing ERβ plus PSMD2 or ERβ plus UBA7. An empty expression vector was also cotransfected into HeLa cells with ERβ expression vector as the control. The overexpression of PSMD2 decreased ERβ protein levels by 2.5-fold in HeLa cells compared with the empty vector control (Fig. 4C). However, the overexpression of UBA7 protein did not degrade ERβ protein compared to the empty vector control (Fig. 4C). Therefore, elevation of PSMD2 could be associated with degradation of ERβ protein in endometriotic lesions by bufalin treatment. To determine the specificity of PSMD2-mediated ERβ degradation, mammalian expression vectors for ERα, full-length SRC-1 and SRC-1 isoform were also cotransfected into HeLa cells along with expression vectors for PSMD2 and UBA7. In contrast with ERβ, however, overexpression of both PSMD2 and UBA7 did not result in the degradation of ERα and SRC-1 isoform proteins (Figs. 4D and 4E). In contrast to the SRC-1 isoform, however, the full-length SRC-1 was degraded by PSMD2 but not by UBA7 (Fig. 4F). Therefore, bufalin-mediated degradation of the full-length SRC-1 might be associated with elevation of PSMD2 in endometriotic lesions. Collectively, these results imply that bufalin disrupts the SRC-1 isoform/ERβ axis in endometriotic lesions by degrading ERβ protein through the elevation of PSMD2.
Bufalin treatment induced apoptosis in epithelial cells but reduced the proliferative activity in stromal cells from endometriotic lesions
To determine the impact of bufalin-induced disruption of the SRC-1 isoform/ERβ axis in endometriosis progression, we examined the alteration of proliferation and apoptosis signaling in endometriotic lesions treated with bufalin compared to the vehicle because hyperproliferation and anti-apoptosis are closely associated with endometriosis progression and the SRC-1 isoform/ERβ axis is involved in dysregulation of apoptosis and proliferation (Han et al. 2012; Han et al. 2015; Pellegrini, et al. 2012; Salmassi, et al. 2011). To determine the proliferative activity in endometriotic lesions, Ki-67 levels were determined by IHC (Fig. 5A). Proliferation in the epithelial compartment of endometriotic lesions was not significantly reduced by bufalin treatment (Fig. 5B). However, bufalin reduced the levels of Ki-67 by 62.5% (P=0.0032) in the stromal compartment of endometriotic lesions compared to the vehicle (Fig. 5C). Therefore, the bufalin treatment significantly reduced the proliferation of endometrial stromal cells but not that of epithelial cells from endometriotic lesions.
Figure 5. Bufalin induced the apoptosis and reduced the proliferation in endometriotic lesions.
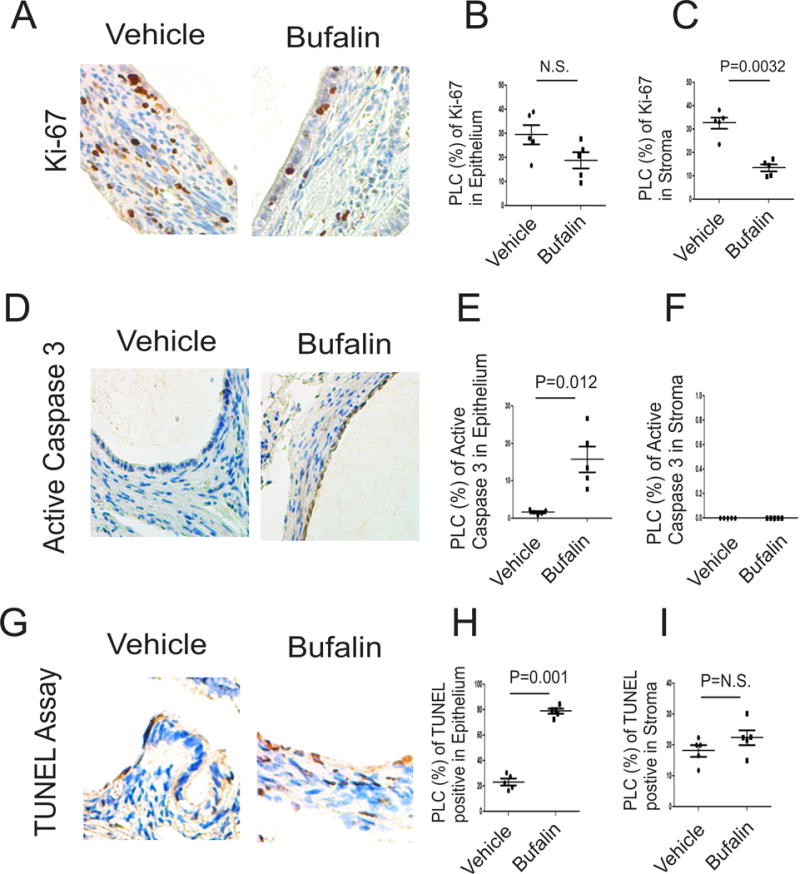
(A–C) Levels of Ki-67 in endometriotic lesions isolated from mice with endometriosis treated with bufalin (1 mg/kg) and vehicle using IHC (A). The levels of Ki67 in epithelial (B) and stromal cells (C) from endometriotic lesions in panel A were quantified by ImageJ (n=5 mice for each group). (D–F) Levels of the active form of caspase 3 protein (D) in epithelial (E) and stromal cells (F) from endometriotic lesions treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=5 mice for each group). (G–I) The number of TUNEL-positive cells (G) in epithelial (H) and stromal cells (I) from endometriotic lesions treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=5 mice for each group). Data are presented as the means ± SEM and P value (Student’s t-test).
In addition to proliferation, we next examined apoptosis signaling by determining the levels of the active form of caspase 3 (Fig. 5D). Bufalin treatment increased the levels of the active form of caspase 3 by 9.8-fold (p=0.012) in epithelial cells from endometriotic lesions compared to the vehicle (Fig. 5E). In contrast with epithelial cells, however, the active form of caspase 3 was not detected in stromal cells in endometriotic lesions treated with bufalin and vehicle (Fig. 5F). In addition to the active form of caspase3, the TUNEL assay also revealed that compared with the vehicle, the bufalin treatment increased the number of TUNEL-positive cells in epithelial cells of the endometriotic lesions (Figs. 5G and 5H). In contrast with epithelial cells, however, bufalin, compared with the vehicle, did not elevate TUNEL-positive cells in stromal cells of endometriotic lesions (Fig. 5I). Collectively, bufalin treatment is associated with elevation of apoptosis signaling in epithelial cells and reduction of proliferation of stromal cells from endometriotic lesions.
Bufalin treatment induced the pyroptosis signaling in stromal cells of endometriotic lesions
In addition to antiapoptosis, alteration of inflammatory signaling has an essential role in endometriosis progression. Therefore, we next examined whether inflammatory signaling is altered in endometriotic lesions by bufalin treatment. Interestingly, bufalin treatment, compared with the vehicle, elevated the active form of IL-1β by 5.1-fold (p=0.0001) in stromal cells but not in epithelial cells (Figs. 6A, 6B and 6C). The elevation of the active form of IL-1β is associated with the progression of pyroptosis (Ying and Padanilam 2016), and caspase I is associated with progression of pyroptosis induced by the active form of IL-1β (Miao, et al. 2011). Therefore, we next determined the levels of caspase I in endometriotic lesions treated with bufalin versus vehicle. In addition to the active form of IL-1β, IHC revealed that the levels of caspase 1 were elevated by 2.9-fold (p=0.0015) in stromal cells, but not in epithelial cells, from bufalin-treated endometriotic lesions compared to the vehicle-treated lesions (Figs. 6D, 6E and 6F). Therefore, bufalin treatment might be associated with elevated signaling of the active form of IL-1β by activating the pyroptosis signaling in stromal compartments of endometriotic lesions.
Figure 6. Bufalin induced the pyroptosis signaling in endometriotic lesions.
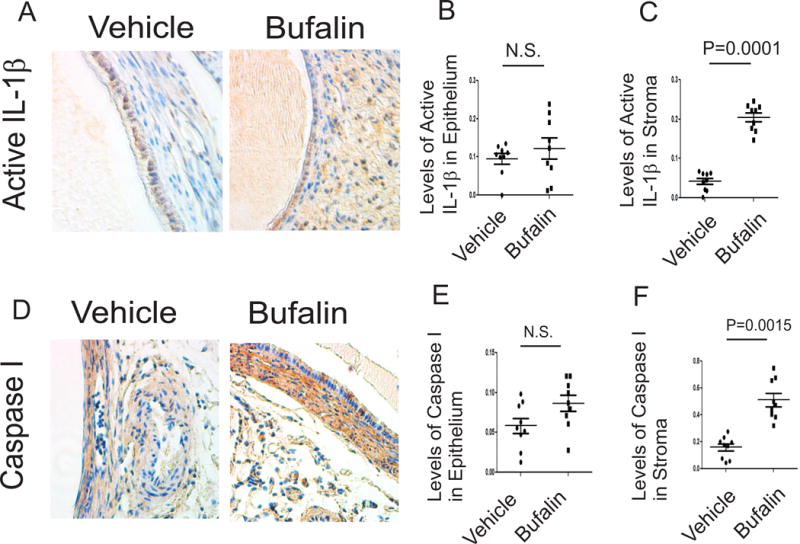
(A–C) Levels of the active form of IL-1β in endometriotic lesions isolated from mice with endometriosis treated with bufalin (1 mg/kg) and vehicle using IHC (A). The levels of the active form of IL-1β in epithelial (B) and stromal cells (C) from endometriotic lesions in panel A were quantified by ImageJ (n=3 mice for each group, three independent IHC assays for each mouse). (D–F) Levels of caspase 1 protein (D) in epithelial (E) and stromal cells (F) from endometriotic lesions treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=3 mice for each group, three independent IHC assays for each mouse). Data are presented as the means ± SEM and P value (Student’s t-test).
Bufalin treatment induced endoplasm reticulum (ER) stress in endometriotic lesions
Pyroptosis elevates the levels of the active form of IL-1β, and then the increased active form of IL-1β promotes ER stress signaling (Liu, et al. 2015; Verma and Datta 2010). Based on these observations, we examined the levels of ER-stress markers, such as PKR-like ER kinase (PERK), protein disulfide isomerase (PDI) and binding immunoglobulin protein (BiP), in endometriotic lesions treated with bufalin (Oslowski and Urano 2011). Compared to the vehicle, bufalin treatment elevated the expression levels of PERK by 9.1-fold (p=0.0001) in epithelial cells from endometriotic lesions (Figs. 7A and 7B). In addition to the epithelial compartment, bufalin also increased PERK levels in stromal cells from endometriotic lesions by 4.1-fold (P=0.0001) (Fig. 7C).
Figure 7. Bufalin induced ER stress signaling in endometriotic lesions.
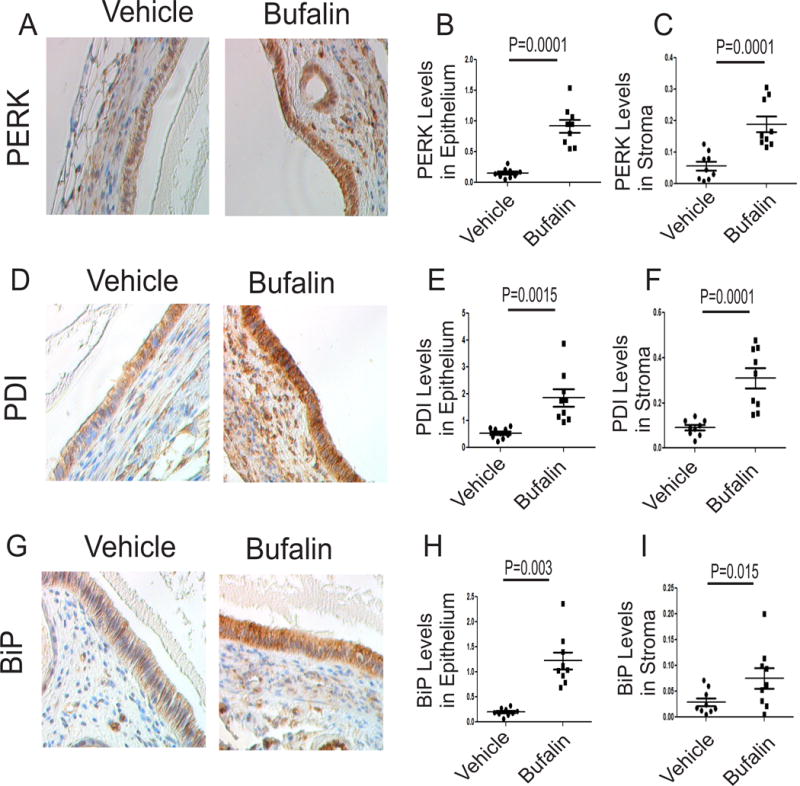
(A–C) Levels of PERK protein (A) in epithelial (B) and stromal cells (C) from endometriotic lesions treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=3 mice for each group, three independent IHC assays for each mouse). (D–F) Levels of PDI protein (D) in epithelial (E) and stromal cells (F) from endometriotic lesions treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=3 mice for each group, three independent IHC assays for each mouse). (G–I) Levels of BiP protein (G) in epithelial (H) and stromal cells (I) from endometriotic lesions treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=3 mice for each group, three independent IHC assays for each mouse). Data are presented as the means ± SEM and P value (Student’s t-test).
The PDI levels were also elevated in epithelial cells (by 4.2-fold, P=0.0015) and stromal cells (by 2.9-fold, P=0.0001) in endometriotic lesions treated with bufalin compared with the vehicle (Figs. 7D, 7E, and 7F). Levels of BiP were elevated by 4.6-fold (p=0.003) in epithelial cells from endometriotic lesions treated with bufalin compared with the vehicle (Figs. 7G and 7H). The bufalin treatment also elevated BiP levels (by 2.1-fold, P=0.015) in stromal cells from endometriotic lesions by bufalin compared with the vehicle (Fig. 7I). Therefore, bufalin treatment mainly stimulates ER-stress signaling in endometriotic lesions.
Bufalin treatment did not impair normal uterine function of mice without endometriosis
Our observations revealed that bufalin treatment elevated apoptosis, pyroptosis and ER stress signaling and reduced the proliferation in endometriotic lesions. These observations raised the question, what is the effect of bufalin in normal uteri? To address this question, normal C57BL/6J mice were treated with bufalin (1 mg/kg for 21 days) and vehicle as the control. IHC with Ki-67 antibody revealed that compared to the vehicle, the bufalin treatment did not reduce the proliferation activity in uteri (Fig. 8A). In addition to proliferation, the level of the active form of caspase 3 was not elevated in uteri by bufalin treatment compared to the vehicle (Fig. 8B). IHC with the active form of IL-1β also revealed that the pyroptosis signaling was not elevated in uteri by bufalin treatment compared to the vehicle (Fig. 8 C).
Figure 8. Bufalin did not impair the normal uterine function.
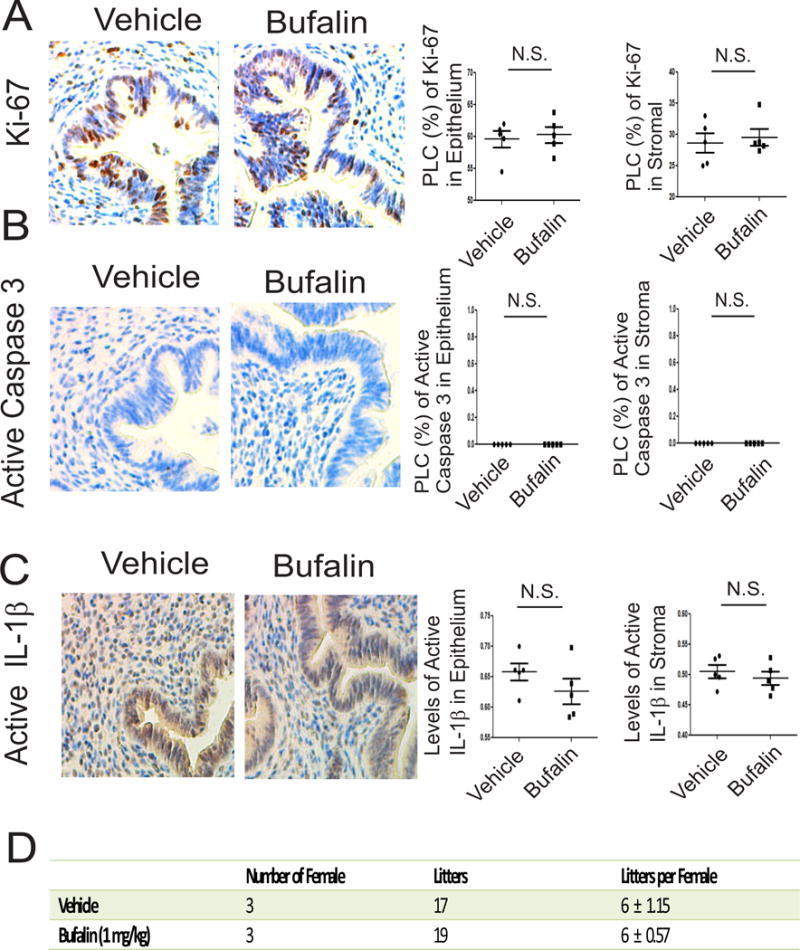
(A) Levels of Ki-67 in endometriotic lesions isolated from mice treated with bufalin (1 mg/kg) and vehicle using IHC. The levels of Ki-67 in epithelial and stromal cells from uteri in panel A were quantified by ImageJ (n=5 mice for each group). (B) Levels of the active form of caspase 3 protein in epithelial and stromal cells from uteri treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=5 mice for each group). (C) Levels of the active form of IL-1β in epithelial and stromal cells from uteri treated with bufalin (1 mg/kg) or vehicle were determined using IHC and then quantified using ImageJ (n=5 mice for each group). (D) The litter size per C57BL/6J female mouse was determined after 21-day treatment with bufalin (1 mg/kg) and vehicle. Data are presented as the means ± SEM and P value (Student’s t-test).
To further validate the effect of bufalin on normal uterine function, we determined the fertility of female mice treated with bufalin (1 mg/kg) for 21 days. The female mouse fertility assay revealed that compared with vehicle treatment, bufalin treatment did not reduce the reproductive activity in female mice (Fig. 8D). In our hands, therefore, the 21-day bufalin treatment (1 mg/kg) did not disrupt the fertility of female mice.
Working model for bufalin-mediated endometriosis suppression
Based on our results, we propose a model for bufalin-induced suppression of endometriosis progression (Fig. 9). Bufalin degrades ERβ protein by elevating PSMD2 and hyperactivates SRC-1 isoform function in endometriotic lesions. This disruption of the bufalin-induced SRC-1 isoform/ERβ axis is associated with activation of apoptosis signaling in epithelial cells and reduces the proliferation of stromal cells. In addition, bufalin treatment stimulates the pyroptosis in stromal cells and then elevates ER stress signaling in endometriotic lesions. These multiple cellular dysregulations by bufalin treatment in endometriotic lesions cause the suppression of endometriosis progression.
Figure 9. The working model for bufalin-mediated suppression of endometriosis progression.
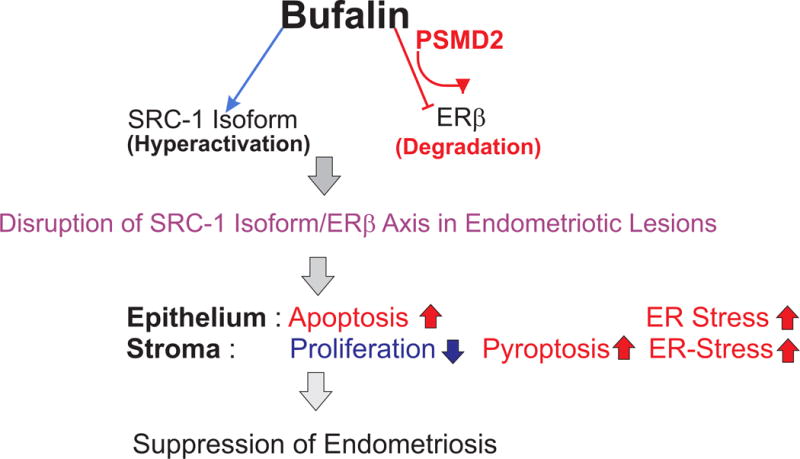
Bufalin hyperactivated the SRC-1 isoform and degraded ERβ in endometriotic lesion to disrupt the SRC-1 isoform/ERβ axis. This disruption of the SRC-1 isoform/ERβ axis stimulated apoptosis in epithelial cells and reduced proliferation in stromal cells of endometriotic lesions. In addition, bufalin activated pyroptosis in stromal cells and enhanced ER stress in endometriotic lesions. The alteration of these cellular pathways should suppress endometriosis progression by bufalin.
Conclusion
Bufalin has been known as a traditional oriental medicine and used for cancer treatment because it induces apoptosis in cancer cells (Takai, et al. 2012). In addition to cancer cells, bufalin also induces apoptosis and G0/G1 cell cycle arrest in endometriotic stromal cells in vitro (Nasu, et al. 2005). However, more detailed studies have not been conducted to evaluate whether bufalin can be employed as an alternative medicine for endometriosis treatment. Bufalin inhibits SRC-1 and SRC-3 functions by degrading their protein levels and inhibiting their intrinsic transcriptional activities in various cancer cells (Wang et al. 2014). However, bufalin hyperactivates the SRC-1 isoform function, unlike that of full-length SRC-1, in endometriotic lesions. Since the SRC-1 isoform has an essential role in endometriosis progression, further activation of the SRC-1 isoform in endometriotic lesions might stimulate endometriosis progression. However, the bufalin-induced hyperactivation of the SRC-1 isoform activity suppressed the growth of endometriotic lesions compared to that of the vehicle. How we can address this discrepancy? The first explanation is that the molecular effects of bufalin on the SRC-1 isoform are quite similar to those of another newly discovered small molecule named MCB613. MCB613 was identified as an SRC small molecule stimulator because treatment with MCB613 stimulated the transcriptional activity of SRC and markedly induced ER stress coupled with the generation of reactive oxygen species to suppress the growth of cancer cells (Wang, et al. 2015). Therefore, overstimulating the SRC oncogenic program can be an effective strategy to kill cancer cells. In the same context, therefore, over-stimulating the endometriosis-promoting SRC-1 isoform using bufalin could kill endometriotic lesions by promoting ER stress induced by pyroptosis, similar to MCB613. Therefore, bufalin is an activator of the SRC-1 isoform, whereas it acts as an inhibitor against full-length SRC-1. The second explanation is that bufalin treatment degrades ERβ in endometriotic lesions. Our previous study revealed that overexpression of the SRC-1 isoform in IHEECs prevented TNFα-induced apoptosis because IHEECs express ERβ (Han et al. 2012). Similar to SRC-1 isoform overexpression, bufalin treatment increased the SRC-1 isoform level and its transcriptional activity in endometriotic lesions. However, bufalin treatment suppressed the growth of endometriotic lesions because ERβ levels were reduced in endometriotic lesions. To prevent apoptosis in endometriotic lesions, both the SRC-1 isoform and ERβ axis are required because the SRC-1 isoform/ERβ complex interacts with the apoptosis machinery in endometriotic lesions (Han et al. 2015). Even though bufalin elevated SRC-1 isoform function in endometriotic lesions, however, bufalin did not prevent apoptosis in endometriotic lesions without ERβ and then reactivated apoptosis signaling.
In addition to the SRC-1 isoform, bufalin treatment also impaired endometriosis-stimulating ERβ signaling in endometriotic lesions by degrading ERβ protein. ERβ has been known as a key driver along with the SRC-1 isoform for endometriosis progression by preventing TNFβ-induced apoptosis and activating inflammasomes (Han et al. 2015; Monsivais, et al. 2014). Therefore, ERβ should be the best target to suppress endometriosis progression because the deactivation of ERβ could reactivate TNFα-induced apoptosis signaling only in endometriotic lesions to suppress their growth. Therefore, bufalin significantly reduced ERβ levels in endometriotic lesions and then reactivated apoptosis signaling in epithelial cells from endometriotic lesions to suppress the endometriosis progression. To degrade ERβ protein, bufalin treatment elevated levels of PSMD2 in endometriotic lesions. PSMD2 is a component of the 19S regulatory component and is responsible for substrate recognition and binding. Interestingly, the elevation of PSMD2 levels drives the progression of various human diseases. In lung cancer, for example, elevated levels of PSMD2 and its gene signature are associated with acquisition of the metastatic phenotype and a poor prognosis because knockdown of PSMD2 decreases proteasome activity and induces growth inhibition and apoptosis in lung cancer cell lines (Matsuyama, et al. 2011). Consistent with the cancer progression, proteasome components also have a crucial role in endometriosis progression because the proteasome inhibitor bortezomib suppresses endometriosis progression in a rat endometriosis model (Celik, et al. 2008). In contrast with this finding, however, the bufalin-induced elevation of PSMD2 suppresses the growth of endometriotic lesions. In addition to survival functions, the proteasome also has an essential role in cell death signaling (Vacca, et al. 2007). In endometriotic tissues, therefore, PSMD2 specifically recognizes ERβ, an essential endometriosis driver, and then degrades it to suppress the ERβ-regulated gene signature that is required for endometriosis progression. Interestingly, PSMD2 did not degrade ERα protein. ERα has critical roles in estrogen target tissues in addition to endometriosis progression, and estrogen deficiency affects different tissues, resulting in an increase in various diseases such as osteoporosis or cardiovascular diseases (Valera, et al. 2015). The side effects of current endometriosis treatment are partly due to the inhibition of ERα signaling in these estrogen target tissues. Therefore, the bufalin/PSMD2/ERβ pathway should improve the specificity of endometriosis treatment and reduce side effects of the inhibition of ERα signaling by the current estrogen depletion therapy.
The disruption of the SRC-1 isoform/ERβ axis by bufalin stimulates both pyroptosis and apoptosis signaling in endometriotic lesions. At first, this observation confused us because two different types of cell death signaling occurred in endometriotic lesions at the same time. However, IHC analyses revealed that the two different cell death signaling pathways are detected in different cellular compartments of endometriotic lesions. For example, activation of pyroptosis and apoptosis are detected in stromal and epithelial cells of endometriotic lesions, respectively. Pyroptosis, or caspase 1-dependent cell death, is known as inflammatory cell death signaling and is initiated by various pathological stimuli, such as stroke, heart attack or cancer (Bergsbaken, et al. 2009). However, the role of pyroptosis in endometriosis progression has not been reported. Here, we revealed that bufalin induced pyroptosis in the stromal compartments of endometriotic lesions by elevating the active form of IL-1β and caspase 1 in stromal cells. IL-1β signaling has been known as a double-edged sword because the elevation of the active form of IL-1β results in proliferative activity in certain cases but also induces cell death signaling in other cases (Kolb, et al. 2014). For survival, for example, ERβ activated caspase 1 activity in inflammasomes and then increased the active form of IL-1β levels in endometriotic lesions to activate cell adhesion and proliferative activity in ectopic lesions (Han et al. 2015). However, bufalin treatment hyperelevated the levels of the active form of IL-β in the stromal compartments of endometriotic lesions compared to the vehicle-treated endometriotic lesions. Hyperactivating IL-1β signaling induced ER stresses, resulting in cell death in human pancreatic cells (Verma and Datta 2010). Therefore, bufalin treatment elevates hyper-releasing active forms of IL-1β from pyroptotic stromal cells and enhances ER stress in endometriotic lesions to effectively suppress the growth of endometriotic lesions along with activation of apoptosis in epithelial cells of endometriotic lesions.
It appears that bufalin itself may not be an ideal drug for endometriosis. Bufalin belongs to the bufadienolide group, and bufadienolides are known for having deleterious cardiovascular side effects due to their inhibition of the transport enzyme Na+/K+-adenosine triphosphatase (Xu, et al. 2016). Despite this known cytotoxic property of bufadienolides, however, they have been used to treat cardiovascular and kidney diseases (Puschett, et al. 2010). Chronic treatment with a low dose of bufadienolides has been proven to be effective in certain instances, causing little to no side effects (Jing, et al. 1994; Panesar 1992). Our study also revealed that 21-day bufalin treatment (1 mg/kg) did not impair normal uterine function and fertility. We know, however, that bufalin has potential cardiac ion channel toxicity so it is not an ideal candidate drug with which to go forward. Although the combination therapy of low-dose bufalin and other drugs could be further investigated, this study mainly points to the value of searching in the future for additional new chemicals other than bufalin that inhibit SRC function as potential therapies for endometriosis.
Acknowledgments
SJH led the entire project. SJH and BWO evaluated all data. SJH, YJC, JL and MJP designed and performed the experiments. SJH, YJC and JL wrote the manuscript. Yeon Jean Cho and Jiyeun Lee equally contributed to this work.
Financial Support: This work was supported by grants from the US National Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, R01HD082786 and R01HD008188 to BWO), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, U24 DK097748 Pilot Grant to SJH) and Mike Hogg Foundation to SJH.
Abbreviations
- BiP
binding immunoglobulin
- COX-2
cyclooxygenase-2
- ER
endoplasmic reticulum
- ERα
estrogen receptor alpha
- ERβ
estrogen receptor beta
- IHEECs
immortalized human endometrial epithelial cells
- IL
interleukin
- PDI
protein disulfide isomerase
- PGE2
prostaglandin E2
- PERK
PKR-like ER kinase
- PSMD2
proteasome 26S subunit, non-ATPase 2
- SMI
small molecular inhibitor
- SRC
steroid receptor coactivator
- UBA7
ubiquitin-like modifier-activating enzyme7
Footnotes
Competing Financial Interests: The authors declare no competing financial interests.
References
- Bedaiwy MA, Alfaraj S, Yong P, Casper R. New developments in the medical treatment of endometriosis. Fertil Steril. 2017;107:555–565. doi: 10.1016/j.fertnstert.2016.12.025. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bono Y, Kyo S, Takakura M, Maida Y, Mizumoto Y, Nakamura M, Nomura K, Kiyono T, Inoue M. Creation of immortalised epithelial cells from ovarian endometrioma. Br J Cancer. 2012;106:1205–1213. doi: 10.1038/bjc.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brilhante AV, Augusto KL, Portela MC, Sucupira LC, Oliveira LA, Pouchaim AJ, Nobrega LR, Magalhaes TF, Sobreira LR. Endometriosis and Ovarian Cancer: an Integrative Review (Endometriosis and Ovarian Cancer) Asian Pac J Cancer Prev. 2017;18:11–16. doi: 10.22034/APJCP.2017.18.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulun SE. Endometriosis. N Engl J Med. 2009;360:268–279. doi: 10.1056/NEJMra0804690. [DOI] [PubMed] [Google Scholar]
- Bulun SE, Cheng YH, Pavone ME, Yin P, Imir G, Utsunomiya H, Thung S, Xue Q, Marsh EE, Tokunaga H, et al. 17Beta-hydroxysteroid dehydrogenase-2 deficiency and progesterone resistance in endometriosis. Semin Reprod Med. 2010;28:44–50. doi: 10.1055/s-0029-1242992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik O, Hascalik S, Elter K, Tagluk ME, Gurates B, Aydin NE. Combating endometriosis by blocking proteasome and nuclear factor-kappaB pathways. Hum Reprod. 2008;23:2458–2465. doi: 10.1093/humrep/den246. [DOI] [PubMed] [Google Scholar]
- Cummings AM, Metcalf JL. Induction of endometriosis in mice: a new model sensitive to estrogen. Reprod Toxicol. 1995;9:233–238. doi: 10.1016/0890-6238(95)00004-t. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Bartley J, David M. Aromatase inhibitors and cyclooxygenase-2 (COX-2) inhibitors in endometriosis: new questions–old answers? Eur J Obstet Gynecol Reprod Biol. 2005;122:144–150. doi: 10.1016/j.ejogrb.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Farland LV, Lorrain S, Missmer SA, Dartois L, Cervenka I, Savoye I, Mesrine S, Boutron-Ruault MC, Kvaskoff M. Endometriosis and the risk of skin cancer: a prospective cohort study. Cancer Causes Control. 2017;28:1011–1019. doi: 10.1007/s10552-017-0939-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farland LV, Tamimi RM, Eliassen AH, Spiegelman D, Hankinson SE, Chen WY, Missmer SA. Laparoscopically Confirmed Endometriosis and Breast Cancer in the Nurses’ Health Study II. Obstet Gynecol. 2016;128:1025–1031. doi: 10.1097/AOG.0000000000001684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenka L, George M, Sen M. A peek into the drug development scenario of endometriosis – A systematic review. Biomed Pharmacother. 2017;90:575–585. doi: 10.1016/j.biopha.2017.03.092. [DOI] [PubMed] [Google Scholar]
- Han SJ, Hawkins SM, Begum K, Jung SY, Kovanci E, Qin J, Lydon JP, DeMayo FJ, O’Malley BW. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat Med. 2012;18:1102–1111. doi: 10.1038/nm.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SJ, Jeong J, Demayo FJ, Xu J, Tsai SY, Tsai MJ, O’Malley BW. Dynamic cell type specificity of SRC-1 coactivator in modulating uterine progesterone receptor function in mice. Mol Cell Biol. 2005;25:8150–8165. doi: 10.1128/MCB.25.18.8150-8165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SJ, Jung SY, Wu S, Hawkins SM, Park MJ, Kyo S, Qin J, Lydon JP, Tsai SY, Tsai MJ, et al. Estrogen Receptor β modulates apoptosis complexes and the inflammasome to drive the pathogenesis of endometriosis. Cell. 2015;163:960–974. doi: 10.1016/j.cell.2015.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata T, Osuga Y, Takamura M, Saito A, Hasegawa A, Koga K, Yoshino O, Hirota Y, Harada M, Taketani Y. Interleukin-17F increases the secretion of interleukin-8 and the expression of cyclooxygenase 2 in endometriosis. Fertil Steril. 2011;96:113–117. doi: 10.1016/j.fertnstert.2011.04.060. [DOI] [PubMed] [Google Scholar]
- Jing Y, Ohizumi H, Kawazoe N, Hashimoto S, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. Selective inhibitory effect of bufalin on growth of human tumor cells in vitro: association with the induction of apoptosis in leukemia HL-60 cells. Jpn J Cancer Res. 1994;85:645–651. doi: 10.1111/j.1349-7006.1994.tb02408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5:12–20. doi: 10.1007/s13238-013-0001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krikun G, Mor G, Alvero A, Guller S, Schatz F, Sapi E, Rahman M, Caze R, Qumsiyeh M, Lockwood CJ. A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology. 2004;145:2291–2296. doi: 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
- Kyo S, Nakamura M, Kiyono T, Maida Y, Kanaya T, Tanaka M, Yatabe N, Inoue M. Successful immortalization of endometrial glandular cells with normal structural and functional characteristics. Am J Pathol. 2003;163:2259–2269. doi: 10.1016/S0002-9440(10)63583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamp M, Peters M, Reinmaa E, Haller-Kikkatalo K, Kaart T, Kadastik U, Karro H, Metspalu A, Salumets A. Polymorphisms in ESR1, ESR2 and HSD17B1 genes are associated with fertility status in endometriosis. Gynecol Endocrinol. 2010 doi: 10.3109/09513590.2010.495434. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhao N, Zhu H, Zhu S, Pan S, Xu J, Zhang X, Zhang Y, Wang J. Circulating interleukin-1beta promotes endoplasmic reticulum stress-induced myocytes apoptosis in diabetic cardiomyopathy via interleukin-1 receptor-associated kinase-2. Cardiovasc Diabetol. 2015;14:125. doi: 10.1186/s12933-015-0288-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama Y, Suzuki M, Arima C, Huang QM, Tomida S, Takeuchi T, Sugiyama R, Itoh Y, Yatabe Y, Goto H, et al. Proteasomal non-catalytic subunit PSMD2 as a potential therapeutic target in association with various clinicopathologic features in lung adenocarcinomas. Mol Carcinog. 2011;50:301–309. doi: 10.1002/mc.20632. [DOI] [PubMed] [Google Scholar]
- McLean AC, Valenzuela N, Fai S, Bennett SA. Performing vaginal lavage, crystal violet staining, and vaginal cytological evaluation for mouse estrous cycle staging identification. J Vis Exp. 2012:e4389. doi: 10.3791/4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243:206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsivais D, Dyson MT, Yin P, Coon JS, Navarro A, Feng G, Malpani SS, Ono M, Ercan CM, Wei JJ, et al. ERbeta- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis. Mol Endocrinol. 2014;28:1304–1315. doi: 10.1210/me.2013-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasu K, Nishida M, Ueda T, Takai N, Bing S, Narahara H, Miyakawa I. Bufalin induces apoptosis and the G0/G1 cell cycle arrest of endometriotic stromal cells: a promising agent for the treatment of endometriosis. Mol Hum Reprod. 2005;11:817–823. doi: 10.1093/molehr/gah249. [DOI] [PubMed] [Google Scholar]
- Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011;490:71–92. doi: 10.1016/B978-0-12-385114-7.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panesar NS. Bufalin and unidentified substance(s) in traditional Chinese medicine cross-react in commercial digoxin assay. Clin Chem. 1992;38:2155–2156. [PubMed] [Google Scholar]
- Pellegrini C, Gori I, Achtari C, Hornung D, Chardonnens E, Wunder D, Fiche M, Canny GO. The expression of estrogen receptors as well as GREB1, c-MYC, and cyclin D1, estrogen-regulated genes implicated in proliferation, is increased in peritoneal endometriosis. Fertil Steril. 2012;98:1200–1208. doi: 10.1016/j.fertnstert.2012.06.056. [DOI] [PubMed] [Google Scholar]
- Poole EM, Lin WT, Kvaskoff M, De Vivo I, Terry KL, Missmer SA. Endometriosis and risk of ovarian and endometrial cancers in a large prospective cohort of U.S. nurses. Cancer Causes Control. 2017;28:437–445. doi: 10.1007/s10552-017-0856-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puschett JB, Agunanne E, Uddin MN. Emerging role of the bufadienolides in cardiovascular and kidney diseases. Am J Kidney Dis. 2010;56:359–370. doi: 10.1053/j.ajkd.2010.01.023. [DOI] [PubMed] [Google Scholar]
- Sacco K, Portelli M, Pollacco J, Schembri-Wismayer P, Calleja-Agius J. The role of prostaglandin E2 in endometriosis. Gynecol Endocrinol. 2012;28:134–138. doi: 10.3109/09513590.2011.588753. [DOI] [PubMed] [Google Scholar]
- Saji S, Jensen EV, Nilsson S, Rylander T, Warner M, Gustafsson JA. Estrogen receptors alpha and beta in the rodent mammary gland. Proc Natl Acad Sci U S A. 2000;97:337–342. doi: 10.1073/pnas.97.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmassi A, Acar-Perk B, Schmutzler AG, Koch K, Pungel F, Jonat W, Mettler L. Apoptosis resistance in endometriosis. Bioimpacts. 2011;1:129–134. doi: 10.5681/bi.2011.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai N, Kira N, Ishii T, Yoshida T, Nishida M, Nishida Y, Nasu K, Narahara H. Bufalin, a traditional oriental medicine, induces apoptosis in human cancer cells. Asian Pac J Cancer Prev. 2012;13:399–402. doi: 10.7314/apjcp.2012.13.1.399. [DOI] [PubMed] [Google Scholar]
- Vacca RA, Valenti D, Bobba A, de Pinto MC, Merafina RS, De Gara L, Passarella S, Marra E. Proteasome function is required for activation of programmed cell death in heat shocked tobacco Bright-Yellow 2 cells. FEBS Lett. 2007;581:917–922. doi: 10.1016/j.febslet.2007.01.071. [DOI] [PubMed] [Google Scholar]
- Valera MC, Gourdy P, Tremollieres F, Arnal JF. From the Women’s Health Initiative to the combination of estrogen and selective estrogen receptor modulators to avoid progestin addition. Maturitas. 2015;82:274–277. doi: 10.1016/j.maturitas.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Vercellini P, Vigano P, Somigliana E, Fedele L. Endometriosis: pathogenesis and treatment. Nat Rev Endocrinol. 2014;10:261–275. doi: 10.1038/nrendo.2013.255. [DOI] [PubMed] [Google Scholar]
- Verma G, Datta M. IL-1beta induces ER stress in a JNK dependent manner that determines cell death in human pancreatic epithelial MIA PaCa-2 cells. Apoptosis. 2010;15:864–876. doi: 10.1007/s10495-010-0498-4. [DOI] [PubMed] [Google Scholar]
- Wang L, Yu Y, Chow DC, Yan F, Hsu CC, Stossi F, Mancini MA, Palzkill T, Liao L, Zhou S, et al. Characterization of a Steroid Receptor Coactivator Small Molecule Stimulator that Overstimulates Cancer Cells and Leads to Cell Stress and Death. Cancer Cell. 2015;28:240–252. doi: 10.1016/j.ccell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O’Malley BW. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol. 2011;25:2041–2053. doi: 10.1210/me.2011-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 2014;74:1506–1517. doi: 10.1158/0008-5472.CAN-13-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Liu X, Schwarz S, Hu L, Guo D, Gu Q, Schwarz W. Inhibitory efficacy of bufadienolides on Na(+), K(+)-pump activity versus cell proliferation. Biochem Biophys Rep. 2016;6:158–164. doi: 10.1016/j.bbrep.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Y, Padanilam BJ. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol Life Sci. 2016;73:2309–2324. doi: 10.1007/s00018-016-2202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Yang YK, Wu WZ. Bufalin attenuates the stage and metastatic potential of hepatocellular carcinoma in nude mice. J Transl Med. 2014;12:57. doi: 10.1186/1479-5876-12-57. [DOI] [PMC free article] [PubMed] [Google Scholar]