

Abstract
Incentive salience, negative emotionality, and executive function are functional domains that are etiologic in the initiation and progression of addictive disorders, having been implicated in humans with addictive disorders and in animal models of addictions. Measures of these three neuroscience-based functional domains can capture much of the effects of inheritance and early exposures that lead to trait vulnerability shared across different addictive disorders. For specific addictive disorders, these measures can be supplemented by agent specific measures such as those that access pharmacodynamic and pharmacokinetic variation attributable to agent-specific gatekeeper molecules including receptors and drug-metabolizing enzymes. Herein, we focus on the translation and reverse translation of knowledge derived from animal models of addiction to the human condition via measures of neurobiological processes that are orthologous in animals and humans, and that are shared in addictions to different agents. Based on preclinical data and human studies, measures of these domains in a general framework of an Addictions Neuroclinical Assessment (ANA) can transform the assessment and nosology of addictive disorders, and can be informative for staging disease progression. We consider next steps and challenges for implementation of ANA in clinical care and research.
1. Introduction
Psychoactive agents that are widely used tend to be widely abused. On a global basis, alcohol, nicotine, and cannabis are widely used, although quantity and qualitative aspects of use vary enormously across time and space, leading to disparities in impact on public health. On an overall basis, and for every population studied, the public health impact of addictive disorders is large. For example, alcohol is consumed worldwide, even in countries where it is illegal to sell or buy it. Correspondingly, alcohol is one of the largest preventable contributors to disease (1), 4.6% of the global disease burden being attributable to alcohol, as measured in disability-adjusted life years. In the United States, roughly 14% of individuals are diagnosable with an alcohol use disorder (AUD) in a given year, and the lifetime prevalence rate of AUD is 29% (2). For several addictive disorders, including AUD, FDA-approved medications are available, and behavioral treatments are also helpful. Treatments for addictive disorders are partially effective, probably helping at least a third of patients who receive them, but treatment is usually not received: over 90% of individuals with AUD never receive specialized treatment (3). The treatment of addictions as end stage diseases in which neuroadaptive changes of addiction, and other damage to the body predominate, and may be difficult to undo, is inherently less satisfactory than prevention, and may be less efficacious than treatments targeted to specific types of vulnerability and stages of progression. However, developing new clinical approaches to addictions based on the neuroscience of addiction – a precision medicine of addictions – ultimately will require integration of relevant neuroscience-based measures into nosology.
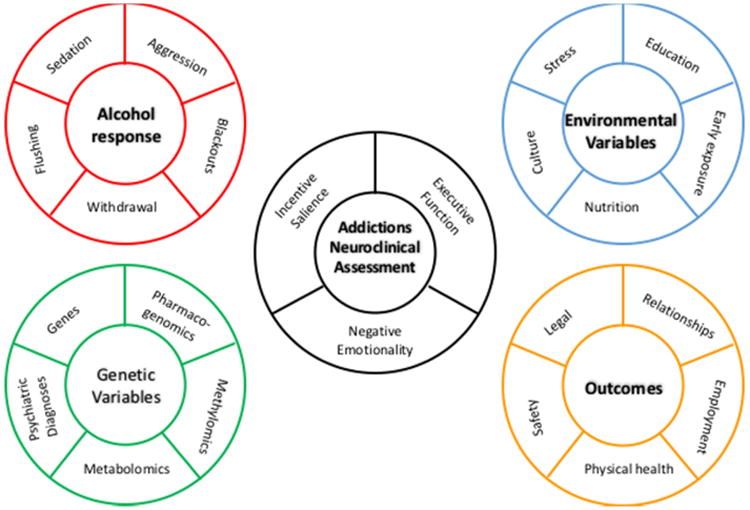
Advances in our functional understanding of the pathophysiology of addictions are derived from studies in humans but also by investigations in animal models that capture different aspects of addictions. However, translating findings from animal models to humans, and reverse translating from humans to animal models, is hampered by the etiologic heterogeneity of addiction vulnerability in both humans and the diversity of animal models available (4). In humans, there is wide variation in progression and outcome, and limited ability to stage addiction using measures of progression emergent from studies in animal models. Animal models of addiction afford a degree of control of exposure and genotype, and access to tissue that is impossible to attain in human studies. Neuroadaptive processes (5), and to a lesser extent, genes, gene transcripts, and proteins (6), have primarily been identified in rodents, with translation to human. Translational studies at the genetic and neuroscience level reveal that mechanisms of addiction in mice, rats, and humans are orthologous, i.e., functionally similar and of a shared evolutionary origin. The roles of both genetic and environmental factors in interindividual variation in vulnerability and progression can bet traced in both humans and animal models of addiction. In rodents, genetically very similar, or identical (inbred and inbred F1) individuals can be divergent in behaviors such as novelty seeking and novelty-induced hyperlocomotion that predict addiction-like behavior, and, by extension, divergent in outcome (4), and divergent in addiction behaviors that develop subsequent to exposure. Preclinical studies are therefore an avenue to identify domains that are critical for predicting liability and staging response in human patients, provided relevant domains can be measured. Herein we link critical findings from animal models of addiction in humans to three neuroscience domains comprising the Addictions Neuroclinical Assessment (ANA) (7), Figure 1. While ANA focuses on addictions broadly, we have elected to focus on alcohol as an exemplar substance of abuse, with additional literature from other substances integrated throughout the manuscript as relevant.
Figure 1. Addictions Neuroclinical Assessment Framework and Ancillary Measures.
1.1 Clinical heterogeneity
Clinical heterogeneity is the major barrier to the treatment of addictive disorders and development of better treatments. In both the Diagnostic and Statistical Manual of Mental Disorders (DSM) and International Classification of Diseases (ICD), addictions are categorical diagnoses based on symptom counts of up to eleven intercorrelated symptoms, with a minimum of two in whatever of (11 × 10)/2 = 55 combinations to meet a threshold diagnosis of DSM addictive disorder. DSM-5 (8) estimates level of severity, also based on symptom counts. Because a diagnosis of addictive disorder under ICD and DSM criteria requires that the patient meet a limited number of criteria from a larger list of partially inter-correlated criteria, there is considerable within-diagnosis heterogeneity. Any patient can reach the endpoints represented by these behaviorally focused criteria via different destinations, and beginning from distinctly different, and even polar opposite, starting points of vulnerability. For example, both anxiety (internalizing behavior: enhanced affective response, prior sensitization to stress/trauma) and risk-taking (externalizing behavior: impulsivity, enhanced responses to novelty, novelty seeking) can predispose to addiction, liability thus arising from genetic risk factors, and exposures. The diagnostic criteria for AUD and other addictive disorders focus on overt behavioral symptoms and consequences of use rather than underlying neurobiological differences that lead to vulnerability and can define progression.
1.2 Development of research diagnostic measures, and criteria
Given the significant advances in our understanding of the neurocircuitry of addiction, e.g., (5), and the neurobehavioral differences that are involved in to vulnerability, and the capacity to measure the activity and output of the brain for relevant domains, the time may be ripe for leveraging knowledge towards a more nuanced approach to diagnosis and treatment. Such an effort would align with similar initiatives in mental health, e.g., the Research Domain Criteria (RDoC) program at the National Institute of Mental Health, and in medicine more generally, e.g., the NIH Precision Medicine Initiative Cohort Program http://www.nih.gov/about-nih/who-we-are/nih-director/statements/preparing-launch-precision-medicine-initiative-cohort-program). One major distinction between ANA and these initiatives is that ANA is focused within a particular disease category, i.e., addiction, rather than across various diseases.
1.3 Traits mediating vulnerability
Historically, most preclinical studies of addiction, including alcohol, have focused on drug reinforcement (4). However, studies in rats and mice have revealed powerful predictors of interindividual variation in liability. Several of these measures correspond to heritable, disease associated intermediate phenotypes (endophenotypes) (9) in people (10). These include trait measures in the anxiety (internalizing) and impulsive-like (externalizing) domains that are the basis of a substantial portion of the quantitative inheritance of addictive disorders (11). While an ability to recognize broad differences in externalizing and internalizing behavior has advanced genetic analyses, and has improved translational alcoholism research, better measures of these domains are needed.
1.4 Shared and unshared liability
People vary in their preference for particular addictive agents both because of a shared vulnerability component and because of “unshared” – agent-specific- components of inheritance (12). For example, alcohol and nicotine use disorders are substantially cross-inherited, as was learned by measuring the sharing of risk for a different disorder by twin siblings of index patients, in large, epidemiologically representative twin samples such as the WWII Veterans, Vietnam Veterans, and Virginia Twin Studies. Opioid Use Disorder was not observed to be as strongly cross-inherited with other addictive disorders (Goldman and Bergen, 1998).
Substance-specific genes influencing addiction include ones whose actions are specific to the substance, or relatively so, and for example ALDH Glu487Lys (relatively specific to AUD, via alcohol-induced flushing) and CYP2A6 variants (relatively specific to nicotine, via nicotine metabolism). Shared liability genes influencing addiction include ones whose actions are shared across different substances such as DAT and HTR2B, whose variants can increase impulsivity, as well as genes such as NPY and FKBP5, whose variants alter emotionality and stress response (13), potentially increasing or decreasing vulnerability to different addictions, and as also influenced by stress exposures.
Unique variations in exposures to addictive agents themselves (availability) can dictate variation in expression of liability is expressed by abuse of one addictive agent or another, or whether liability is penetrant at all. For example, a drug such as Khat is used nearly universally in certain countries, e.g., those in East Africa and Arabia, but is virtually unavailable in others, e.g., European countries without sizable East African immigrants, and rarely used (14). For any addictive agent, agent-specific measures are required to assess level and pattern of consumption, and to index response. For example, carbohydrate deficient transferrin (CDT) and plasma sialic acid index of apolipoprotein A for alcohol (15) and cotinine and 3′-hydroxycotinine for nicotine (16) and hair (17) and blood (18) levels of cannabis can be used to augment history reports, such as the Timeline Follow-Back (19).
Agent-specific responses to drug challenges can index aspects of vulnerability that are relatively agent-specific, serving as bioassays in the absence of genotypes or biochemical measures that might be used as predictive or explanatory surrogates. For AUD, responses that have been found to be predictive, and that have been correspondingly been traced in both animal models humans, include initial sedative sensitivity to sedative/hypnotic drugs such as alcohol (20), stimulant and sedative responses to alcohol (21), withdrawal severity, and motor activation. The latter predicts shared vulnerability to addiction to different agents. Motor activation occurs on the ascending limb of the blood alcohol concentration curve when drinking is initiated, but is also triggered by novelty, by drugs such as cocaine that release dopamine, and by drugs that act directly on dopamine receptors. Shared sedative sensitivity between alcohol and benzodiazepine drugs may also underpin a portion of the shared sensitivity to alcohol-induced sedation, and as supported by genetic mapping studies in rodents (22), by preliminary genetic studies in people identifying GABAA6 as a potential shared liability locus for alcohol and benzodiazepine effect on eye movements (23), and by cross-sedation and cross-tolerance effects of alcohol and other sedative hypnotic drugs. However, genes, and processes critical to sedative response to alcohol would not be likely to be shared with stimulant drugs such as amphetamine and cocaine, or with gambling. Tolerance to the sedative effects of alcohol that can be mediated by induction of metabolism by cytochrome P450 enzyme activity (pharmacokinetic tolerance) and by neural resilience (pharmacodynamics) can be agent-specific, or specific to a class of agents sharing pharmacologic or physiologic mode of action. Capture of agent-specific responses has an important place in defining vulnerability, and progression.
However, whereas agent-specific assessments need to be developed, the broader need is to assess common domains of vulnerability that lead to cross-inheritance (24), and to common pathways in outcome. The rationale for ANA is therefore both to subclassify patients addicted to particular agents, reducing diagnostic heterogeneity, but also to measure dimensions that act across agent-based diagnostic boundaries, and that can improve convergence between preclinical and clinical models. Here, we review prior classification attempts for AUD, a representative addictive disorder for which efforts to develop a classification schemes has been most intensive, noting that these schemes are primarily focused on a single addictive agent. We describe the stages of the addiction cycle and how they relate to the neuroscience domains for ANA. Next we review evidence for disruption in these domains from animals and humans associated with addiction. We build on the description of ANA in (7), but specifically focus on linkage and consilience between animal and human findings in AUD.
2. Clinically-based classification of AUD
Subclassification of alcohol addiction began with Jellinek (25), and was continued by Cloninger (26), who identified the Type I versus Type II dichotomy that has become well-known, i.e., Type I patients who are characterized by relatively later onset of addiction, a combination of genetic and environmental mediators, and differing levels of severity, and Type II patients who have early onset, and who tend to have stronger genetic liability to alcohol addiction, and more severe clinical presentation. The clinical classification schemes were important in demonstrating the existence of heterogeneity, but the Cloninger typology also proposed a neurobiological basis of vulnerability grounded in three heritable dimensions of personality: novelty seeking, harm avoidance and reward dependence, posited to be related to the functions of particular neurochemical circuits, and later perseverance. Although later findings have been inconsistent with respect to these dimensions and their applicability to addictions (27), the Cloninger typology represented a starting point in reconceptualizing addictive disorders in terms of combinations of characteristics within a three-dimensional space, the combinations of those characteristics being specific to individuals. Additional efforts included those by Babor (28) and Lesch (29). Later, latent class analysis was applied, e.g., (30), (31). These schemes mostly used demographic and alcohol use characteristics to subdivide AUD. Identified groups varied by age of onset, gender, family history, and clinical course. Absent data related to the neurobiology of addiction, however, these attempts were unable to clarify the mechanisms underlying alcoholism.
In this context, Cloninger was the first to propose an RDoC-like framework to measure factors underlying mental disorders. Cloninger's index traits, which can be measured via the Tridimensional Personality Questionnaire (TPQ) (32), substantially correspond to major personality traits previously extracted by factor analysis of questionnaire data, and for example harm avoidance roughly equates to neuroticism, as measured on the Eysenck Scale (33). This correlation does not diminish the TPQ as a scale-based measurement of personality adapted to AUD, but points to the need to measure functions that are more directly tied to vulnerability and progression of addiction, some of which substantially overlap with personality traits. Recent advances in the neuroscience of addiction coupled with technological breakthroughs in neuroimaging and genomics afford the ability to use these molecular and machine data and questionnaire and performance-based measures that most strongly track the neurobiological changes related to addiction (34, 35). Consistent with the intention of the TPQ, ANA focuses on brain physiology and measures tracking addiction-relevant aspects of vulnerability and outcome (Figure 1).
3. Stages of the Addiction Cycle and the Addictions Neuroclinical Assessment
Addiction does not occur immediately on exposure to a psychoactive agent, but develops over time. The addiction cycle may be understood from various perspectives, including social psychological; psychiatric, dysadaptational, and neurobiologic. In its totality, addiction is an allostatic process, meaning a transition to a new, more dysfunctional state that is then defended. The dynamic, progressive nature of the addictive process is a fundamental aspect of addiction that has been missing in schemes that have attempted to subclassify addictions. For example, patients with AUD, across populations, are more anxious (harm avoidant) regardless of whether they are of the early onset type, with higher impulsivity and novelty seeking, or whether they were of the late-onset type, in whom initial predisposition is more likely to be in the internalizing domain of increased anxiety and emotional (affective) response (36).
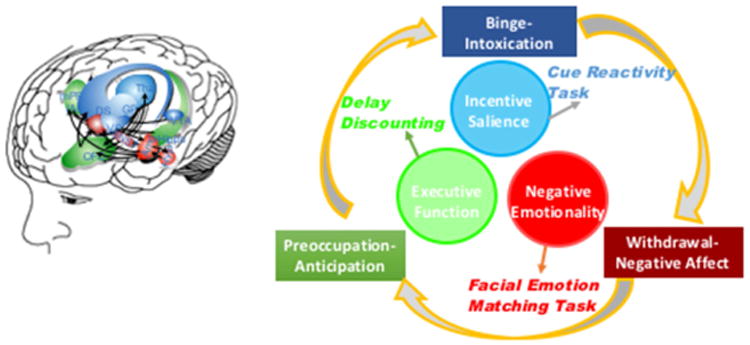
ANA can assess three domains corresponding to phases in the cycle of addiction: binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation (37). At each stage a particular neuroscience domain is most salient: incentive salience with binge/intoxication, negative emotionality with withdrawal/negative affect, and executive function with preoccupation/anticipation. The relationships between the stages and domains, including relevant neurocircuitry and example tasks, appear in Figure 2. There is enormous individual variability in length of time spent in a given phase of the cycle and in the importance of domains of the cycle for different people and different drugs. In addition, little is known about the extent of the individual variation in these domains, or of the progression or reversibility of these changes, in people with addictive disorders.
Figure 2. Addiction Cycle and Relevant Neurocircuitry.
3.1 Incentive Salience
Through repeated use of alcohol and other addictive agents, cues associated with the agent become salient. In the laboratory, preclinical models using alcohol self-administration with varying schedules of reinforcement have traced this shift from goal-directed to habitual behavior. As alcohol cues become incentivized, animals shift from goal-tracking, i.e., pursuit of a particular goal, such as consumption of a rewarding drug, to sign-tracking, i.e., pursuit of the cues they have learned to associate with the goal (38). Thus, the habits related to alcohol consumption themselves become an incentive for other behavior, a phenomenon that has been termed “incentive habit” (39). At the neural level, these changes manifest in the basal ganglia and related structures, as demonstrated in progression from basolateral amygdala-ventral striatal interactions to central amygdala-dorsal striatal connections (40) in reward responding. Cellular-molecular changes involved in the habit-engaging plasticity of the basal ganglia include glutamate receptors, proteins involved in dendritic spines and nerve growth factors (41). Related and critical lines of work link shifts in dopamine receptor expression to the development of incentive salience. Preclinical studies suggest that decreased expression of the dopamine D2 receptor enhances alcohol intake both in animals trained to self-administer ethanol and in non-alcohol preferring rats (42). The changes in incentive salience are likely due to altered connectivity and firing of neuronal ensembles that span multiple brain regions with a key focus on the neurocircuitry of the basal ganglia (43), and in which many molecules of different classes including endocannabinoids (44), monoamine neurotransmitters, and neuropeptides play important roles. Preclinical work in rats has also identified causal roles for neuronal ensembles in the central nucleus of the amygdala (45) and the infralimbic cortex (46), in contributing to excessive alcohol consumption and seeking, respectively, in rats.
The neurocircuitry associated with the development of incentive salience includes structures within the basal ganglia (5). When the ventral striatum is activated, striatal-pallidal-thalamocortical loops are engaged and then involve the dorsal striatum; these circuits underlie the development of habit formation (40). Relevant synaptic alterations include alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and glutamate-modulated N-methyl-D-aspartate (NMDA) receptors in projections from the PFC and amygdala to the ventral tegmental area and nucleus accumbens (47-49), and changes to the metabotropic glutamate receptor subtype 2 (mGluR2), such that mGluR2 loss in corticoaccumbal circuitry may lead to increased risk for relapse (50). As conditioned cues engage these circuits, the patient progresses through the addiction cycle; these circuits also mediate drug craving and compulsive use upon exposure to cues and/or stress that constitute behavioral expressions of addiction. In multiple ways, progression to addiction involves dopamine receptors, including (a) changes in tonic dopamine cell firing resulting in lower, stable dopamine levels in projections to the PFC, largely mediated by D1 receptors (51, 52); and (b) inhibition of cyclic adenosine monophosphate (cAMP) signalling by D2 receptors (53, 54). Data also suggests a role for D3 receptors, which are co-located in the nucleus accumbens with D1 receptors (55).
Increased incentive salience of drug-related stimuli is observed both in addicted and at-risk individuals. In individuals addicted to alcohol, presentation of alcohol cues induces craving for alcohol and is predictive of relapse (56, 57). Treatment aimed at extinguishing cue reactivity to alcohol reduced fMRI activation in the left ventral striatum, an area associated with reward, in the treatment group compared to controls (58). In an example of convergence between animal and human addiction models, correlation between decreased D2 expression and drug preference in rodents is paralleled by positron emission tomography (PET) findings of decreased D2 receptor availability in striatum among individuals addicted to cocaine (59) and alcohol (60) as well as increased D2 availability in striatum in non-alcoholic family members of alcoholics (61). However, research using postmortem brains from alcoholics found alterations in D1 and DA transporter binding, but not D2 (62). Further, abstinent individuals with AUDs have increased activation of the ventral striatum in response to alcohol cue presentation, an association not found among individuals without alcohol addiction (63). Similarly, heavy drinking individuals without an AUD diagnosis showed increased BOLD activation in the bilateral insula and left ventral striatum to alcohol cues when compared to light drinking individuals (64). These data suggest recruitment of incentive salience-reward networks accompanying addiction and heavy use. Finally, a meta-analysis including 176 studies identified common neural pathways for both natural rewards, e.g., food, sex, and drug cue-induced craving, implying that the development of incentive salience relates to neural networks associated with processing for non-drug rewards, affect, and memory (65).
What is less clear is what drives whether some individuals are predisposed to developing increased salience for alcohol-related cues. Potentially, people at greater risk may find natural rewards less rewarding, and therefore experience drugs as more potent reinforcers. Individuals with a positive family history had increased neural activation to alcohol cues compared to family history negative individuals (66). Natural variation in reward, adaptive in other circumstances, can predispose the individual to addiction by requiring more powerful reinforcers, e.g., a psychoactive drug than those ordinarily offered by food or social companionship.
3.2 Negative Emotionality
Negative affective states, including dysphoria, anhedonia, alexithymia, and anxiety, are all associated with alcohol addiction, particularly during withdrawal and craving. Individuals with AUD demonstrate increased negative emotional responses to alcohol-related stimuli, along with higher levels of overall low mood (67, 68). The “self-medication” or “tension-reduction” theories of excessive alcohol consumption have support in both clinical practice and research findings through relief of negative affective states, i.e., negative reinforcement (37, 69). Reduced hedonic capacity is well-recognized as a clinical feature of AUD (70-72) and is associated with increased craving for alcohol (73), see above. This finding emphasizes the complex relationship between negative affect and increased alcohol cue reactivity among those addicted to alcohol.
One of the major molecules hypothesized to underlie these negative emotional states are the brain stress systems including corticotropin-releasing factor (CRF) (74). As addiction progresses, upregulation of neural stress systems, including CRF, occurs in parallel with the downregulated neural reward systems (e.g., those in the basal ganglia), as described above (75), and some brain stress systems such as the dynorphin-kappa system may directly decrease reward function (76). For example, alcohol-dependent rats were more sensitive to CRF and CRF1 antagonists with respect to the increase and decrease, respectively of release of GABA in CeA interneurons (54). Further, administration of CRF1 antagonists reduced alcohol consumption in both alcohol-dependent and –naïve animals (77). Valdez and colleagues found similar evidence of a role for CRF in mediating anxiety-like behaviors and alcohol self-administration in dependent rats (78). Dynorphin-kappa antagonists can block excessive drinking associated with dependence in rodent models and would be hypothesized to reverse the decrease in dopamine function produced by activation of brain dynorphin (76). Similar findings on the relevance of upregulated stress systems for alcohol addiction are reviewed in (79) and in consideration of their relevance for translation into treatments for humans in (67).
Neural circuitry associated with negative emotionality primarily comprises areas involving the ventral striatum, extended amygdala, and lateral habenula (5). Decreased dopaminergic and serotonergic transmission in the nucleus accumbens during acute withdrawal (80), and decreased GABAergic and increased NMDA glutamatergic transmission, also in the nucleus accumbens (81, 82), demonstrate alterations in ventral striatum circuitry. Within the extended amygdala, CRF, norepinephrine, and dynorphin are all recruited and underlie the negative affect manifested in withdrawal (83, 84). Finally, the lateral habenula facilitates the experience of unpleasant states and is thus involved in mediating negative affect (85). The habenula regulates reductions in dopamine neuron firing in the ventral tegmental area; these reductions mediate the inability to accept an anticipated reward (86, 87).
The development of compulsive drug seeking may be closely related to negative emotionality, given that compulsive drug-seeking is strongly associated with negative, rather than positive affect, the “dark side” of addiction (88). However, incentive salience and impulsivity driven by negative emotions also may have particular relevance for AUD (89), given the noted increases in various forms of impulsivity in this population. Moreover, negative emotional responses to stress are strongly linked to increased risk for relapse (90). The high comorbidity between mood and anxiety disorders with AUD provide further evidence for increased levels of negative affect among individuals with AUD. An assortment of functional polymorphisms, some, such as variants in FKBP5 and NPY, altering functions of genes in the HPA and brain stress axes and others, such as variants at SLC6A4 and COMT, altering functions of downstream targets, can predispose individuals to be more or less emotionally responsive and stress responsive (13). However, levels of negative emotionality may vary considerably depending on the stage of the addiction cycle. As already pointed out earlier, patients with AUD tend to be anxious, approximately one standard deviation higher in Harm Avoidance, regardless of their initial predisposition (36). Neuroscience-based measures of negative emotionality include response time and attention paid to negative vs. neutral or positive cues, i.e., negative attentional bias. Individuals with an AUD show a greater BOLD response to negative pictures compared to individuals without an AUD (91). Further, limbic activity associated with negative emotional stimuli was increased in individuals with AUD and this activity was associated with increases in craving in AUD individuals and absent in healthy volunteers (92). Similarly, personalized stress and neutral scripts hypoactivated prefrontal cortex and hyperactivated insula and ventral striatum in patients with AUD, as reviewed in (93). Together, these data suggest that individuals with AUD tend to be hyperreactive to stimuli associated with negative emotions but have hypoactivate circuits that regulate emotion, i.e. prefrontal regions, leading to emotion dysregulation and contributing to relapse.
Given the robust associations between CRF, stress, and increased alcohol intake among animals, the findings that CRF antagonism did not reduce stress-induced alcohol craving in humans (94, 95) were surprising, but suggests a more complex association between these systems in humans than in rodents. Note, however, that a vasopressin 1B antagonist did show efficacy in promoting abstinence particularly in subjects showing baseline high stress (96). Indeed, these examples illustrate the challenges inherent in translational alcohol research: that the models used in rodent research may not have direct analogs in human research, or that additional processes beyond direct neurobiology likely mediate links between CRF, stress, and alcohol craving. Individual differences in CRF genotype may also influence these responses (97).
3.3 Executive Function
The domain of executive function encompasses planning, working memory, attention, response inhibition, decision-making, set-shifting, and cognitive flexibility. Impairment in executive function which is often directly attributable to dysfunction of prefrontal cortex plays a role in many psychiatric illnesses, including addiction.
People with AUD often manifest impaired executive functions. In addition to the original data reviewed below, we also direct attention to recent comprehensive reviews on alcoholism and executive function, e.g., (98-100). Case-control studies show that executive cognitive impairments are part of the natural history of AUD. Patients with AUD show reduced executive functioning via multiple metrics, including neuropsychological tests and neuroimaging measures. Patients with AUD perform more poorly on tests of spatial working memory, with evidence to suggest that they activate different neural circuits in different ways to perform the same task (101) and in line with the cortical insufficiency hypothesis of Goldman-Rakic (102). Similarly, a study of verbal working memory found no differences in behavioral outcomes between those with AUD and those without, but there were different patterns of neural activation between the two groups (103). One polymorphism COMT Val158Met, that has been associated with AUD, alters frontal cortical efficiency, and presumably under via frontal dopamine mechanism, i.e., increased dopamine in the PFC is associated with improved executive functions, as proposed by Goldman-Rakic (104). Deficits in planning (105), set-shifting (106), and response inhibition (107) are more common in individuals with AUD than without. Relatedly, impulsivity was correlated with reduced reward anticipation in the ventral striatum among alcoholics but not in non-alcoholic comparison individuals (108), suggesting that high impulsivity is related to difficulty maintaining positive expectations about rewards. Finally, a recent meta-analysis of executive function impairments in AUD found some of the most significant executive impairments were in the domains of planning, response inhibition, and problem-solving (106). There is some recovery of function demonstrated during protracted abstinence (109), although ability to recover may be compromised among older patients (110).
Studies in animal models are beginning to elucidate specific mechanisms for alcohol induced frontal cortex dysfunction. Chronic intermittent ethanol exposure triggers remodeling of neurons in the prefrontal cortex of mice (111). Relatedly, binge drinking in adolescent rats reduced myelin density in the corpus callosum and predicted worse performance on a working memory task in adulthood (112). Behavioral consequences of these ethanol-induced changes in prefrontal circuits includes increased premature responding (measuring a type of impulsivity known as waiting impulsivity) among rodents experiencing alcohol withdrawal (113) and more risky choices made by alcohol-exposed adolescent rats (114). One potential mechanism of action through which PFC dysfunction may lead to excessive drinking is increased expression of Fos+ neurons in the medial PFC, which is associated both with transient impairments in working memory during acute withdrawal and increased alcohol consumption following reinstatement (115). Critically, frontal dysfunction, although discussed here under the executive function domain, relates to disruptions in the domains of negative emotionality and incentive salience, as well, given that the PFC is part of the circuitry mediating those domains (116). There is strong correspondence between the animal data showing a role of the prelimbic prefrontal cortex and anterior cingulate glutamatergic projections to the nucleus accumbens driving drug and cue-induced reinstatement (47) and compelling data in human imaging studies showing that corresponding frontal cortex regions in humans are activated in cue induced craving (117, 118). In imaging studies, cues associated with alcohol, nicotine, cocaine and opioids all show activation of areas of the prefrontal cortex such as dorsal lateral prefrontal cortex and cingulate cortex regions (119). Presumably these same circuits contribute not only to the development of incentive salience but also to craving and relapse. Finally, as reviewed (120), epigenetic mechanisms offer another area of study of the impact of alcohol on prefrontal cortical function, and an additional treatment target.
Impairments in executive functions also increase vulnerability to addiction, as shown both by genetic (twin and family studies, and to a small extent, gene studies) and longitudinal studies including studies of heavy and binge users who do not initially meet criteria. In studies of individuals who are genetically at risk, the evidence is somewhat mixed, with some showing clear evidence of disrupted executive function among offspring of alcoholics, while others show no difference as compared with individuals lacking this family history. For example, (121) found that individuals with family histories of alcohol dependence had poorer executive functioning as assessed by neuropsychological testing and increased impulsivity, and impairments in attention (122), response inhibition (123), and set-shifting (124) were observed in children of alcoholics. In contrast, (125) did not find neuropsychological impairment among men with family histories of AUD compared to men without. Perhaps the genetically transmitted liability in those families was of a different type.
Several genetic polymorphisms and rarer genetic variants have been discovered that affect executive function, including the COMT Val158Met missense variant (126) previously referenced, an HTR2B stop codon common in Finns and absent elsewhere (127) and an MAOA VNTR (128). Discovery of these variants that influence executive function is expected because of variation in executive cognitive performance is heritable. However, addiction to alcohol and other drugs is associated with long-term impairments in executive function, secondary to direct neurotoxicity impairing the circuitry of executive cognition, and possibly secondary to indirect effects, and for example as might be induced by withdrawal or stress. The neurotoxic and cognition-altering effects of alcohol and other drugs are highly variable and modulated by multiple factors including level of exposure and genotype (129). One study (130) found lower working memory and planning scores among binge-drinking college males; the same effects were not found for females. In contrast, others (131) found poorer working memory and increased impulsivity among female binge-drinkers in particular. Deconstruction of the factors driving binge drinking can be aided by assessment of prior drinking history, comorbid drug use, and frequency of binges. Overall, there are clear data to support alterations in executive function processes among individuals with addictive disorders, and evidence to suggest that disruptions in executive functions may be linked to vulnerability, at least in some individuals. As with the other neuroscience domains, it is likely that problems in executive functioning play a larger role in the development and maintenance of AUD and other addictive disorders for some people more than others.
4. Improving Translation and Reverse Translation
As reviewed above, there is considerable evidence for disruptions in three neuroscience domains associated with AUD and other addictions from both preclinical and clinical studies. What is less clear is how well specific experimental tasks and findings translate from animals to humans, e.g., the failure of CRF antagonism to reduce alcohol craving in humans, after robustly attenuating alcohol consumption in rodents. Similarly, the ability to reverse-translate human addiction constructs for study in preclinical models, for example, the abundance of clinical data on executive function difficulty in humans, with limited models (e.g., response inhibition) used in preclinical studies, is hampered by a failure to apply tasks measuring the neurobiology of addiction. For example, modeling negative affect in rodents is often done by measuring (lack of) exploration of novel environments, which likely does not adequately represent the complexity of human negative affective states. The importance of individual differences in liability makes the need for better translation and reverse translation all the more pressing, as these individual differences (e.g., in genotype or exposure to trauma) may be experimentally controlled in animals, but not in humans. The development of a battery of assessments based on the neurobiology of addiction, which would then be applied to testing individuals along a continuum of misuse of alcohol and other addictive agents, could foster both translational and reverse translational efforts.
As reviewed (4), animal models of addiction have made significant progress towards improved consilience with constructs relevant for addiction. Currently, robust models exist for phenomena including drug self-administration, drug reinstatement (modeling relapse in humans), relapse preceded by abstinence (forced in dependent animals), drug seeking (compulsive and non-compulsive), and loss of control over drug taking. These behavioral assays are complemented by paradigms designed to assess specific vulnerability traits, such as anxiety, novelty-seeking, and impulsivity. While there is some overlap with the ANA constructs, e.g., anxiety with negative emotionality, and impulsivity with executive (dys)function, there is room for improvement. Specifically, models that expand on other aspects of executive control not related to impulsivity, such as attention or planning, would be useful, as would increased breadth of constructs relevant for negative emotionality. Piloting ANA in humans might help identify specific behavioral constructs relevant for addiction, which could then be reverse translated to preclinical research, e.g., as related to specific aspects of executive function or negative emotionality.
5. Final Points
In summary, we have presented a framework for understanding addiction, the Addictions Neuroclinical Assessment, and reviewed the literature supporting the application of the three ANA neuroscience domains to better understanding AUD and other addictions. A common framework of assessment of addiction neurobiology, e.g., as shown in Figure 1, can enable aspects of vulnerability and consequences of addictions that are shared across different addictive agents to be tracked with the same tools, and leading to larger, harmonized datasets, and leading to a better understanding of shared etiology and consequences. The ANA domains are also orthologous to three of the five RDoC domains. Our implementation plan will begin with a research clinical focus before diversifying both samples and scope, recognizing that as a clinical tool any panel of assessments requires validation, and streamlining. Specifically, we plan to implement a battery of measures, including self-report and behavioral, in the NIAAA intramural research program, which includes individuals across the spectrum of alcohol use and misuse. This platform provides an ideal setting for the deep phenotyping of individuals needed to begin implementation and will include collection of ancillary, relevant measures, such as those related to physiologic function, substance use history, psychiatric comorbidities, stress exposure, and personality, among others, including genetics. Collection of this sample will allow for various types of statistical analysis to be performed as necessary, including a refinement of the ANA battery. We will determine next steps once we have completed this initial data collection and analysis. Of critical importance is meshing ANA data with other levels of information, including neuroimaging and genetics. For too long, treatment for AUD (and other addictive disorders) has been driven primarily by patient self-report and clinical observation. While invaluable to guiding treatment both today and in the future, clinical observation is insufficient to the challenge of personalized medicine for addictive disorders, and to exploiting new insights into the neuroscience of addictions.
An additional potential benefit of ANA will be improving the process for medications development for new pharmacotherapies to treat addictive disorders. So far, medications for AUD have shown only limited effectiveness, in part due to the heterogeneity of the disease (132). One approach to increase effectiveness is to improve precision medicine, identifying subpopulations that are most likely to respond to a specific medication, and integrating this information into all phases of drug development (133). ANA could provide information on the different domains to characterize subjects more fully and perhaps, identify moderators of an experimental pharmacotherapy treatment. This would, undoubtedly, improve the effect size of candidate medications, an important long-term goal of advancing medications development during the next decade. ANA could identify new targets for potential therapies, with a focus on, for example, reducing stress-induced craving, executive dysfunction in addiction, or anxiety-related disorders in addiction. The measures used in the ANA battery could themselves be treatment-related outcomes, i.e., treatment efficacy may be assessed by performance on a given behavioral task. There are similar efforts to treat cognitive impairments in schizophrenia (134), with considerations described for clinical trials of schizophrenia in (135). A compound to treat major depressive disorder (MDD), vortioxetine, demonstrated efficacy for treating the cognitive dysfunction that often occurs with MDD (136, 137). These examples from other psychiatric disorders suggest that using outcomes identified through ANA may more precisely target addiction-related phenotypes. Further, behavioral phenotypes identified through ANA may serve as potential moderators of treatment response, and for example the combination of high craving and liking for sweets moderating treatment response to naltrexone (138). Finally, the ANA domains and associated phenotypes may be used in conjunction with pharmacogenetics approaches to target addiction, and suggest a combined pharmacogenetics-behavioral phenotype strategy may improve precision in medications development to treat addictive disorders.
Finally, although we have largely discussed the three neuroscience domains separately, there is significant overlap between them at the levels of causation and observation. For example, attention, an executive function, is directed towards particular stimuli, such as those cues associated with alcohol, and in the context of a particular affective state. Thus, it may be the functional and structural connectivity between regions of these neural circuitries that unravel areas for target through treatment, whether that means strengthening or weakening connections between a given region. Through this knowledge base that is both broad and deep, we may more effectively target existing treatments and develop new ones, all with the aim of alleviating the enormous global health burden attributable to alcohol and other addictive agents.
This review describes three neuroscience domains relevant for addiction
These comprise Incentive Salience, Negative Emotionality and Executive Function
Preclinical and clinical addiction studies show alterations in these domains
The domains can assist translational and reverse translational addictions research
Acknowledgments
We acknowledge the Division of Intramural Clinical and Biological Research, the Office of the Clinical Director, the Office of the Director, the Laboratory of Neurogenetics, and the Division of Medications Development, all at NIAAA.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. The Lancet. 2009;373:2223–2233. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- 2.Grant BF, Goldstein RB, Saha TD, et al. Epidemiology of dsm-5 alcohol use disorder: Results from the national epidemiologic survey on alcohol and related conditions iii. JAMA Psychiatry. 2015;72:757–766. doi: 10.1001/jamapsychiatry.2015.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Services HaH, editor. SAMHSA. 2013 National Survey on Drug Use and Health. Washington, DC: 2013. [Google Scholar]
- 4.Belin D, Belin-Rauscent A, Everitt BJ, Dalley JW. In search of predictive endophenotypes in addiction: insights from preclinical research. Genes, Brain and Behavior. 2016;15:74–88. doi: 10.1111/gbb.12265. [DOI] [PubMed] [Google Scholar]
- 5.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. The Lancet Psychiatry. 2016;3:760–773. doi: 10.1016/S2215-0366(16)00104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Z, Karlsson C, Liang T, Xiong W, Kimura M, Tapocik JD, et al. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proceedings of the National Academy of Sciences. 2013;110:16963–16968. doi: 10.1073/pnas.1309839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwako LE, Momenan R, Litten RZ, Koob GF, Goldman D. Addictions Neuroclinical Assessment: A Neuroscience-Based Framework for Addictive Disorders. Biol Psychiatry. 2016;80:179–189. doi: 10.1016/j.biopsych.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.APA. Diagnostic and statistical manual of mental disorders (DSM-5®) American Psychiatric Pub; 2013. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 10.Kendler K, Neale M. Endophenotype: a conceptual analysis. Molecular psychiatry. 2010;15:789–797. doi: 10.1038/mp.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kendler K, Aggen S, Prescott C, Crabbe J, Neale M. Evidence for multiple genetic factors underlying the DSM-IV criteria for alcohol dependence. Molecular psychiatry. 2012;17:1306–1315. doi: 10.1038/mp.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldman D, Bergen A. General and specific inheritance of substance abuse and alcoholism. Archives of General Psychiatry. 1998;55:964–965. doi: 10.1001/archpsyc.55.11.964. [DOI] [PubMed] [Google Scholar]
- 13.Bevilacqua L, Goldman D. Genetics of emotion. Trends in cognitive sciences. 2011;15:401–408. doi: 10.1016/j.tics.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffiths P, Lopez D, Sedefov R, Gallegos A, Hughes B, Noor A, et al. Khat use and monitoring drug use in Europe: The current situation and issues for the future. Journal of Ethnopharmacology. 2010;132:578–583. doi: 10.1016/j.jep.2010.04.046. [DOI] [PubMed] [Google Scholar]
- 15.Javors MA, Johnson BA. Current status of carbohydrate deficient transferrin, total serum sialic acid, sialic acid index of apolipoprotein J and serum β-hexosaminidase as markers for alcohol consumption. Addiction. 2003;98:45–50. doi: 10.1046/j.1359-6357.2003.00582.x. [DOI] [PubMed] [Google Scholar]
- 16.Strasser AA, Benowitz NL, Pinto AG, Tang KZ, Hecht SS, Carmella SG, et al. Nicotine Metabolite Ratio Predicts Smoking Topography and Carcinogen Biomarker Level. Cancer Epidemiology Biomarkers & Prevention. 2011;20:234–238. doi: 10.1158/1055-9965.EPI-10-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilkins D, Haughey H, Cone E, Huestis M, Foltz R, Rollins D. Quantitative analysis of THC, 11-OH-THC, and THCCOOH in human hair by negative ion chemical ionization mass spectrometry. Journal of analytical toxicology. 1995;19:483–491. doi: 10.1093/jat/19.6.483. [DOI] [PubMed] [Google Scholar]
- 18.Lowe RH, Karschner EL, Schwilke EW, Barnes AJ, Huestis MA. Simultaneous quantification of Δ 9-tetrahydrocannabinol, 11-hydroxy-Δ 9-tetrahydrocannabinol, and 11-nor-Δ 9-tetrahydrocannabinol-9-carboxylic acid in human plasma using two-dimensional gas chromatography, cryofocusing, and electron impact-mass spectrometry. Journal of Chromatography A. 2007;1163:318–327. doi: 10.1016/j.chroma.2007.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sobell LC, Sobell MB. Measuring alcohol consumption. Springer; 1992. Timeline follow-back; pp. 41–72. [Google Scholar]
- 20.Schuckit MA. Subjective responses to alcohol in sons of alcoholics and control subjects. Archives of General Psychiatry. 1984;41:879–884. doi: 10.1001/archpsyc.1984.01790200061008. [DOI] [PubMed] [Google Scholar]
- 21.Hendler RA, Ramchandani VA, Gilman J, Hommer DW. Behavioral neurobiology of alcohol addiction. Springer; 2011. Stimulant and sedative effects of alcohol; pp. 489–509. [DOI] [PubMed] [Google Scholar]
- 22.Belknap JK, Metten P, Beckley EH, Crabbe JC. Multivariate analyses reveal common and drug-specific genetic influences on responses to four drugs of abuse. Trends in Pharmacological Sciences. 2008;29:537–543. doi: 10.1016/j.tips.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwata N, Cowley DS, Radel M, Roy-Byrne PP, Goldman D. Relationship between a GABAAα6 Pro385Ser substitution and benzodiazepine sensitivity. American Journal of Psychiatry. 1999 doi: 10.1176/ajp.156.9.1447. [DOI] [PubMed] [Google Scholar]
- 24.Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nature Reviews Genetics. 2005;6:521–532. doi: 10.1038/nrg1635. [DOI] [PubMed] [Google Scholar]
- 25.Bowman KM, Jellinek EM. Alcohol addiction and its treatment. Quarterly Journal of Studies on Alcohol. 1941;2:98–176. [Google Scholar]
- 26.Cloninger C. Neurogenetic adaptive mechanisms. Science. 1987;236:410–416. doi: 10.1126/science.2882604. [DOI] [PubMed] [Google Scholar]
- 27.Howard MO, Kivlahan D, Walker RD. Cloninger's tridimensional theory of personality and psychopathology: applications to substance use disorders. Journal of studies on alcohol. 1997;58:48–66. doi: 10.15288/jsa.1997.58.48. [DOI] [PubMed] [Google Scholar]
- 28.Babor TF, Dolinsky ZS, Meyer RE, Hesselbrock M, Hofmann M, Tennen H. Types of alcoholics: concurrent and predictive validity of some common classification schemes. British journal of addiction. 1992;87:1415–1431. doi: 10.1111/j.1360-0443.1992.tb01921.x. [DOI] [PubMed] [Google Scholar]
- 29.Lesch OM, Walter H. Subtypes of alcoholism and their role in therapy. Alcohol and Alcoholism. 1996;31:63–67. [PubMed] [Google Scholar]
- 30.Moss HB, Chen CM, Yi Hy. Subtypes of alcohol dependence in a nationally representative sample. Drug and alcohol dependence. 2007;91:149–158. doi: 10.1016/j.drugalcdep.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sintov ND, Kendler KS, Young-Wolff KC, Walsh D, Patterson DG, Prescott CA. Empirically defined subtypes of alcohol dependence in an Irish family sample. Drug and alcohol dependence. 2010;107:230–236. doi: 10.1016/j.drugalcdep.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cloninger CR, Svrakic DM, Przybeck TR. A psychobiological model of temperament and character. Archives of general psychiatry. 1993;50:975–990. doi: 10.1001/archpsyc.1993.01820240059008. [DOI] [PubMed] [Google Scholar]
- 33.Zuckerman M, Cloninger CR. Relationships between Cloninger's, Zuckerman's, and Eysenck's dimensions of personality. Personality and Individual differences. 1996;21:283. doi: 10.1016/0191-8869(96)00042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuckit MA, Smith TL, Paulus MP, Tapert SF, Simmons AN, Tolentino NJ, et al. The Ability of Functional Magnetic Resonance Imaging to Predict Heavy Drinking and Alcohol Problems 5 Years Later. Alcoholism: Clinical and Experimental Research. 2016;40:206–213. doi: 10.1111/acer.12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whelan R, Watts R, Orr CA, Althoff RR, Artiges E, Banaschewski T, et al. Neuropsychosocial profiles of current and future adolescent alcohol misusers. Nature. 2014;512:185–189. doi: 10.1038/nature13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ducci F, Enoch MA, Funt S, Virkkunen M, Albaugh B, Goldman D. Increased anxiety and other similarities in temperament of alcoholics with and without antisocial personality disorder across three diverse populations. Alcohol. 2007;41:3–12. doi: 10.1016/j.alcohol.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 38.Srey CS, Maddux JMN, Chaudhri N. The attribution of incentive salience to Pavlovian alcohol cues: a shift from goal-tracking to sign-tracking. Frontiers in behavioral neuroscience. 2015;9 doi: 10.3389/fnbeh.2015.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belin D, Belin-Rauscent A, Murray JE, Everitt BJ. Addiction: failure of control over maladaptive incentive habits. Current Opinion in Neurobiology. 2013;23:564–572. doi: 10.1016/j.conb.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 40.Belin D, Jonkman S, Dickinson A, Robbins TW, Everitt BJ. Parallel and interactive learning processes within the basal ganglia: Relevance for the understanding of addiction. Behavioural Brain Research. 2009;199:89–102. doi: 10.1016/j.bbr.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 41.Ron D, Barak S. Molecular mechanisms underlying alcohol-drinking behaviours. Nature Reviews Neuroscience. 2016 doi: 10.1038/nrn.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thanos PK, Volkow ND, Freimuth P, Umegaki H, Ikari H, Roth G, et al. Overexpression of dopamine D2 receptors reduces alcohol self-administration. Journal of neurochemistry. 2001;78:1094–1103. doi: 10.1046/j.1471-4159.2001.00492.x. [DOI] [PubMed] [Google Scholar]
- 43.Koya E, Uejima JL, Wihbey KA, Bossert JM, Hope BT, Shaham Y. Role of ventral medial prefrontal cortex in incubation of cocaine craving. Neuropharmacology. 2009;56:177–185. doi: 10.1016/j.neuropharm.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caillé S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. The journal of neuroscience. 2007;27:3695–3702. doi: 10.1523/JNEUROSCI.4403-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Guglielmo G, Crawford E, Kim S, Vendruscolo LF, Hope BT, Brennan M, et al. Recruitment of a Neuronal Ensemble in the Central Nucleus of the Amygdala Is Required for Alcohol Dependence. Journal of Neuroscience. 2016;36:9446–9453. doi: 10.1523/JNEUROSCI.1395-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfarr S, Meinhardt MW, Klee ML, Hansson AC, Vengeliene V, Schönig K, et al. Losing control: excessive alcohol seeking after selective inactivation of cue-responsive neurons in the infralimbic cortex. Journal of Neuroscience. 2015;35:10750–10761. doi: 10.1523/JNEUROSCI.0684-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature Reviews Neuroscience. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 48.Lüscher C, Malenka R. Drug-Evoked Synaptic Plasticity in Addiction: From Molecular Changes to Circuit Remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolf ME, Ferrario CR. AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci Biobehav Rev. 2010;35:185–211. doi: 10.1016/j.neubiorev.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meinhardt MW, Hansson AC, Perreau-Lenz S, Bauder-Wenz C, Stählin O, Heilig M, et al. Rescue of infralimbic mGluR2 deficit restores control over drug-seeking behavior in alcohol dependence. Journal of Neuroscience. 2013;33:2794–2806. doi: 10.1523/JNEUROSCI.4062-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grace AA. The tonic/phasic model of dopamine system regulation and its implications for understanding alcohol and psychostimulant craving. Addiction. 2000;95:119–128. doi: 10.1080/09652140050111690. [DOI] [PubMed] [Google Scholar]
- 52.Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 53.Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, et al. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nature neuroscience. 2011;14:1033–1038. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caine SB, Negus SS, Mello NK, Patel S, Bristow L, Kulagowski J, et al. Role of dopamine D2-like receptors in cocaine self-administration: studies with D2 receptor mutant mice and novel D2 receptor antagonists. The Journal of neuroscience. 2002;22:2977–2988. doi: 10.1523/JNEUROSCI.22-07-02977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vorel SR, Ashby CR, Paul M, Liu X, Hayes R, Hagan JJ, et al. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. The Journal of neuroscience. 2002;22:9595–9603. doi: 10.1523/JNEUROSCI.22-21-09595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cooney NL, Litt MD, Morse PA, Bauer LO, Gaupp L. Alcohol cue reactivity, negative-mood reactivity, and relapse in treated alcoholic men. Journal of Abnormal Psychology. 1997;106:243–250. doi: 10.1037//0021-843x.106.2.243. [DOI] [PubMed] [Google Scholar]
- 57.Rohsenow DJ, Monti PM, Rubonis AV, Sirota AD, Niaura RS, Colby SM, et al. Cue reactivity as a predictor of drinking among male alcoholics. Journal of Consulting and Clinical Psychology. 1994;62:620–626. doi: 10.1037//0022-006x.62.3.620. [DOI] [PubMed] [Google Scholar]
- 58.Vollstädt-Klein S, Loeber S, Kirsch M, Bach P, Richter A, Bühler M, et al. Effects of Cue-Exposure Treatment on Neural Cue Reactivity in Alcohol Dependence: A Randomized Trial. Biological Psychiatry. 2011;69:1060–1066. doi: 10.1016/j.biopsych.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 59.Volkow ND, Fowler JS, Wolf AP, Schlyer D, Shiue CY, Alpert R, et al. Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am J Psychiatry. 1990;147:719–724. doi: 10.1176/ajp.147.6.719. [DOI] [PubMed] [Google Scholar]
- 60.Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemann R, Ding YS, et al. Decreases in Dopamine Receptors but not in Dopamine Transporters in Alcoholics. Alcoholism: Clinical and Experimental Research. 1996;20:1594–1598. doi: 10.1111/j.1530-0277.1996.tb05936.x. [DOI] [PubMed] [Google Scholar]
- 61.Volkow ND, Wang G, Begleiter H, et al. High levels of dopamine d2 receptors in unaffected members of alcoholic families: Possible protective factors. Archives of General Psychiatry. 2006;63:999–1008. doi: 10.1001/archpsyc.63.9.999. [DOI] [PubMed] [Google Scholar]
- 62.Hirth N, Meinhardt MW, Noori HR, Salgado H, Torres-Ramirez O, Uhrig S, et al. Convergent evidence from alcohol-dependent humans and rats for a hyperdopaminergic state in protracted abstinence. Proceedings of the National Academy of Sciences. 2016;113:3024–3029. doi: 10.1073/pnas.1506012113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wrase J, Schlagenhauf F, Kienast T, Wüstenberg T, Bermpohl F, Kahnt T, et al. Dysfunction of reward processing correlates with alcohol craving in detoxified alcoholics. Neuroimage. 2007;35:787–794. doi: 10.1016/j.neuroimage.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 64.Ihssen N, Cox WM, Wiggett A, Fadardi JS, Linden DE. Differentiating heavy from light drinkers by neural responses to visual alcohol cues and other motivational stimuli. Cerebral cortex. 2011;21:1408–1415. doi: 10.1093/cercor/bhq220. [DOI] [PubMed] [Google Scholar]
- 65.Noori HR, Linan AC, Spanagel R. Largely overlapping neuronal substrates of reactivity to drug, gambling, food and sexual cues: A comprehensive meta-analysis. European Neuropsychopharmacology. 2016;26:1419–1430. doi: 10.1016/j.euroneuro.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 66.Dager AD, Anderson BM, Stevens MC, Pulido C, Rosen R, Jiantonio-Kelly RE, et al. Influence of Alcohol Use and Family History of Alcoholism on Neural Response to Alcohol Cues in College Drinkers. Alcoholism: Clinical and Experimental Research. 2013;37:E161–E171. doi: 10.1111/j.1530-0277.2012.01879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heilig M, Thorsell A, Sommer WH, Hansson AC, Ramchandani VA, George DT, et al. Translating the neuroscience of alcoholism into clinical treatments: from blocking the buzz to curing the blues. Neuroscience & Biobehavioral Reviews. 2010;35:334–344. doi: 10.1016/j.neubiorev.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sinha R, Fox HC, Hong KA, Bergquist K, Bhagwagar Z, Siedlarz KM. Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharmacology. 2009;34:1198–1208. doi: 10.1038/npp.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker TB, Piper ME, McCarthy DE, Majeskie MR, Fiore MC. Addiction Motivation Reformulated: An Affective Processing Model of Negative Reinforcement. Psychological Review. 2004;111:33–51. doi: 10.1037/0033-295X.111.1.33. [DOI] [PubMed] [Google Scholar]
- 70.Hatzigiakoumis DS, Martinotti G, Giannantonio MD, Janiri L. Anhedonia and substance dependence: clinical correlates and treatment options. Frontiers in psychiatry. 2011;2:10. doi: 10.3389/fpsyt.2011.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heinz A, Schmidt LG, Reischies FM. Anhedonia in schizophrenic, depressed, or alcohol-dependent patients--neurobiological correlates. Pharmacopsychiatry. 1994;27(1):7–10. doi: 10.1055/s-2007-1014317. [DOI] [PubMed] [Google Scholar]
- 72.Martinotti G, Nicola MD, Reina D, Andreoli S, Foca F, Cunniff A, et al. Alcohol protracted withdrawal syndrome: the role of anhedonia. Substance use & misuse. 2008;43:271–284. doi: 10.1080/10826080701202429. [DOI] [PubMed] [Google Scholar]
- 73.Janiri L, Martinotti G, Dario T, Reina D, Paparello F, Pozzi G, et al. Anhedonia and substance-related symptoms in detoxified substance-dependent subjects: a correlation study. Neuropsychobiology. 2005;52:37–44. doi: 10.1159/000086176. [DOI] [PubMed] [Google Scholar]
- 74.Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: A key role in the neurobiology of addiction. Frontiers in Neuroendocrinology. 2014;35:234–244. doi: 10.1016/j.yfrne.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koob GF. The dark side of emotion: the addiction perspective. European journal of pharmacology. 2015;753:73–87. doi: 10.1016/j.ejphar.2014.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chavkin C, Koob GF. Dynorphin, Dysphoria, and Dependence: the Stress of Addiction. Neuropsychopharmacology. 2016;41:373–374. doi: 10.1038/npp.2015.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin Releasing Factor–Induced Amygdala Gamma-Aminobutyric Acid Release Plays a Key Role in Alcohol Dependence. Biological Psychiatry. 2010;67:831–839. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, et al. Increased Ethanol Self-Administration and Anxiety-Like Behavior During Acute Ethanol Withdrawal and Protracted Abstinence: Regulation by Corticotropin-Releasing Factor. Alcoholism: Clinical and Experimental Research. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- 79.Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction. 2006;101:23–30. doi: 10.1111/j.1360-0443.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 80.Weiss F, Markou A, Lorang MT, Koob GF. Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain research. 1992;593:314–318. doi: 10.1016/0006-8993(92)91327-b. [DOI] [PubMed] [Google Scholar]
- 81.Dahchour A, De Witte P, Bolo N, Nédélec JF, Muzet M, Durbin P, et al. Central effects of acamprosate:part 1. Acamprosate blocks the glutamate increase in the nucleus accumbens microdialysate in ethanol withdrawn rats. Psychiatry Research: Neuroimaging. 1998;82:107–114. doi: 10.1016/s0925-4927(98)00016-x. [DOI] [PubMed] [Google Scholar]
- 82.Davidson M, Shanley B, Wilce P. Increased NMDA-induced excitability during ethanol withdrawal: a behavioural and histological study. Brain research. 1995;674:91–96. doi: 10.1016/0006-8993(94)01440-s. [DOI] [PubMed] [Google Scholar]
- 83.Delfs J, Zhu Y, Druhan J, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- 84.Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, et al. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76:370–382. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hikosaka O. The habenula: from stress evasion to value-based decision-making. Nature reviews neuroscience. 2010;11:503–513. doi: 10.1038/nrn2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kimura M, Satoh T, Matsumoto N. What does the habenula tell dopamine neurons? Nature neuroscience. 2007;10:677–678. doi: 10.1038/nn0607-677. [DOI] [PubMed] [Google Scholar]
- 87.Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447:1111–1115. doi: 10.1038/nature05860. [DOI] [PubMed] [Google Scholar]
- 88.Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature neuroscience. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- 89.Verdejo-Garcia A, Bechara A, Recknor EC, Perez-Garcia M. Negative emotion-driven impulsivity predicts substance dependence problems. Drug and alcohol dependence. 2007;91:213–219. doi: 10.1016/j.drugalcdep.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 90.Cooney NL, Litt MD, Morse PA, Bauer LO, Gaupp L. Alcohol cue reactivity, negative-mood reactivity, and relapse in treated alcoholic men. Journal of abnormal psychology. 1997;106:243–250. doi: 10.1037//0021-843x.106.2.243. [DOI] [PubMed] [Google Scholar]
- 91.Gilman JM, Hommer DW. IMAGING STUDY: Modulation of brain response to emotional images by alcohol cues in alcohol-dependent patients. Addiction biology. 2008;13:423–434. doi: 10.1111/j.1369-1600.2008.00111.x. [DOI] [PubMed] [Google Scholar]
- 92.Lee E, Ku J, Jung YC, Lee H, An SK, Kim KR, et al. Neural evidence for emotional involvement in pathological alcohol craving. Alcohol and alcoholism. 2013;48:288–294. doi: 10.1093/alcalc/ags130. [DOI] [PubMed] [Google Scholar]
- 93.Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacology & therapeutics. 2011;129:149–171. doi: 10.1016/j.pharmthera.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kwako LE, Spagnolo PA, Schwandt ML, Thorsell A, George DT, Momenan R, et al. The corticotropin releasing hormone-1 (CRH1) receptor antagonist pexacerfont in alcohol dependence: a randomized controlled experimental medicine study. Neuropsychopharmacology. 2015;40:1053–1063. doi: 10.1038/npp.2014.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schwandt ML, Cortes CR, Kwako LE, George DT, Momenan R, Sinha R, et al. The CRF1 antagonist verucerfont in anxious alcohol dependent women: translation of neuroendocrine, but not of anti-craving effects. Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ryan ML, Falk DE, Fertig JB, Rendenbach-Mueller B, Katz DA, Tracy KA, et al. A Phase 2, Double-Blind, Placebo-Controlled Randomized Trial Assessing the Efficacy of ABT-436, a Novel V1b Receptor Antagonist, for Alcohol Dependence. Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clarke TK, Schumann G. Gene–environment interactions resulting in risk alcohol drinking behaviour are mediated by CRF and CRF1. Pharmacology Biochemistry and Behavior. 2009;93:230–236. doi: 10.1016/j.pbb.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 98.Bickel WK, Jarmolowicz DP, Mueller ET, Gatchalian KM, McClure SM. Are executive function and impulsivity antipodes? A conceptual reconstruction with special reference to addiction. Psychopharmacology. 2012;221:361–387. doi: 10.1007/s00213-012-2689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Montgomery C, Fisk JE, Murphy PN, Ryland I, Hilton J. The effects of heavy social drinking on executive function: a systematic review and meta-analytic study of existing literature and new empirical findings. Human Psychopharmacology: Clinical and Experimental. 2012;27:187–199. doi: 10.1002/hup.1268. [DOI] [PubMed] [Google Scholar]
- 100.Wilcox CE, Dekonenko CJ, Mayer AR, Bogenschutz MP, Turner JA. Cognitive control in alcohol use disorder: deficits and clinical relevance. Reviews in the Neurosciences. 2014;25:1–24. doi: 10.1515/revneuro-2013-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pfefferbaum A, Desmond JE, Galloway C, Menon V, Glover GH, Sullivan EV. Reorganization of Frontal Systems Used by Alcoholics for Spatial Working Memory: An fMRI Study. NeuroImage. 2001;14:7–20. doi: 10.1006/nimg.2001.0785. [DOI] [PubMed] [Google Scholar]
- 102.Williams GV, Rao SG, Goldman-Rakic PS. The physiological role of 5-HT2A receptors in working memory. The Journal of neuroscience. 2002;22:2843–2854. doi: 10.1523/JNEUROSCI.22-07-02843.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Desmond JE, Chen SHA, DeRosa E, Pryor MR, Pfefferbaum A, Sullivan EV. Increased frontocerebellar activation in alcoholics during verbal working memory: an fMRI study. NeuroImage. 2003;19:1510–1520. doi: 10.1016/s1053-8119(03)00102-2. [DOI] [PubMed] [Google Scholar]
- 104.Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proceedings of the National Academy of Sciences. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Joyce EM, Robbins TW. Frontal lobe function in Korsakoff and non- Korsakoff alcoholics: Planning and spatial working memory. Neuropsychologia. 1991;29:709–723. doi: 10.1016/0028-3932(91)90067-i. [DOI] [PubMed] [Google Scholar]
- 106.Stephan RA, Alhassoon OM, Allen KE, Wollman SC, Hall M, Thomas WJ, et al. Meta-analyses of clinical neuropsychological tests of executive dysfunction and impulsivity in alcohol use disorder. The American Journal of Drug and Alcohol Abuse. 2016:1–20. doi: 10.1080/00952990.2016.1206113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Noël X, Bechara A, Dan B, Hanak C, Verbanck P. Response inhibition deficit is involved in poor decision making under risk in nonamnesic individuals with alcoholism. Neuropsychology. 2007;21:778–786. doi: 10.1037/0894-4105.21.6.778. [DOI] [PubMed] [Google Scholar]
- 108.Beck A, Schlagenhauf F, Wüstenberg T, Hein J, Kienast T, Kahnt T, et al. Ventral Striatal Activation During Reward Anticipation Correlates with Impulsivity in Alcoholics. Biological Psychiatry. 2009;66:734–742. doi: 10.1016/j.biopsych.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 109.Pitel AL, Rivier J, Beaunieux H, Vabret F, Desgranges B, Eustache F. Changes in the Episodic Memory and Executive Functions of Abstinent and Relapsed Alcoholics Over a 6-Month Period. Alcoholism: Clinical and Experimental Research. 2009;33:490–498. doi: 10.1111/j.1530-0277.2008.00859.x. [DOI] [PubMed] [Google Scholar]
- 110.Munro CA, Saxton J, Butters MA. The Neuropsychological Consequences of Abstinence Among Older Alcoholics: A Cross-Sectional Study. Alcoholism: Clinical and Experimental Research. 2000;24:1510–1516. [PubMed] [Google Scholar]
- 111.Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PloS one. 2012;7:e37541. doi: 10.1371/journal.pone.0037541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vargas WM, Bengston L, Gilpin NW, Whitcomb BW, Richardson HN. Alcohol binge drinking during adolescence or dependence during adulthood reduces prefrontal myelin in male rats. The Journal of Neuroscience. 2014;34:14777–14782. doi: 10.1523/JNEUROSCI.3189-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Walker SE, Peña-Oliver Y, Stephens DN. Learning not to be impulsive: disruption by experience of alcohol withdrawal. Psychopharmacology. 2011;217:433–442. doi: 10.1007/s00213-011-2298-0. [DOI] [PubMed] [Google Scholar]
- 114.Mejia-Toiber J, Boutros N, Markou A, Semenova S. Impulsive choice and anxiety-like behavior in adult rats exposed to chronic intermittent ethanol during adolescence and adulthood. Behavioural Brain Research. 2014;266:19–28. doi: 10.1016/j.bbr.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, et al. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proceedings of the National Academy of Sciences. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.George O, Koob GF. Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neuroscience & Biobehavioral Reviews. 2010;35:232–247. doi: 10.1016/j.neubiorev.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kober H, Lacadie CM, Wexler BE, Malison RT, Sinha R, Potenza MN. Brain activity during cocaine craving and gambling urges: An fMRI study. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Ding YS, et al. Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. The Journal of neuroscience. 2005;25:3932–3939. doi: 10.1523/JNEUROSCI.0433-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jasinska AJ, Stein EA, Kaiser J, Naumer MJ, Yalachkov Y. Factors modulating neural reactivity to drug cues in addiction: a survey of human neuroimaging studies. Neuroscience & Biobehavioral Reviews. 2014;38:1–16. doi: 10.1016/j.neubiorev.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Heilig M, Barbier E, Johnstone AL, Tapocik J, Meinhardt MW, Pfarr S, et al. Reprogramming of mPFC transcriptome and function in alcohol dependence. Genes, Brain and Behavior. 2016 doi: 10.1111/gbb.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gierski F, Hubsch B, Stefaniak N, Benzerouk F, Cuervo-Lombard C, Bera-Potelle C, et al. Executive Functions in Adult Offspring of Alcohol-Dependent Probands: Toward a Cognitive Endophenotype? Alcoholism: Clinical and Experimental Research. 2013;37:E356–E363. doi: 10.1111/j.1530-0277.2012.01903.x. [DOI] [PubMed] [Google Scholar]
- 122.Poon E, Ellis DA, Fitzgerald HE, Zucker RA. Intellectual, cognitive, and academic performance among sons of alcoholics during the early school years: Differences related to subtypes of familial alcoholism. Alcoholism: Clinical and Experimental Research. 2000;24:1020–1027. [PubMed] [Google Scholar]
- 123.Schweinsburg AD, Paulus MP, Barlett VC, Killeen LA, Caldwell LC, Pulido C, et al. An fMRI Study of Response Inhibition in Youths with a Family History of Alcoholism. Annals of the New York Academy of Sciences. 2004;1021:391–394. doi: 10.1196/annals.1308.050. [DOI] [PubMed] [Google Scholar]
- 124.Corral M, Holguín SR, Cadaveira F. Neuropsychological characteristics of young children from high-density alcoholism families: a three-year follow-up. Journal of studies on alcohol. 2003;64:195–199. doi: 10.15288/jsa.2003.64.195. [DOI] [PubMed] [Google Scholar]
- 125.Gillen R, Hesselbrock V. Cognitive functioning, ASP, and family history of alcoholism in young men at risk for alcoholism. Alcoholism: Clinical and Experimental Research. 1992;16:206–214. doi: 10.1111/j.1530-0277.1992.tb01365.x. [DOI] [PubMed] [Google Scholar]
- 126.Smolka MN, Schumann G, Wrase J, Grüsser SM, Flor H, Mann K, et al. Catechol-O-methyltransferase val158met genotype affects processing of emotional stimuli in the amygdala and prefrontal cortex. The Journal of neuroscience. 2005;25:836–842. doi: 10.1523/JNEUROSCI.1792-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bevilacqua L, Doly S, Kaprio J, Yuan Q, Tikkanen R, Paunio T, et al. A population-specific HTR2B stop codon predisposes to severe impulsivity. Nature. 2010;468:1061–1066. doi: 10.1038/nature09629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- 129.Srivastava V, Buzas B, Momenan R, Oroszi G, Pulay AJ, Enoch MA, et al. Association of SOD2, a mitochondrial antioxidant enzyme, with gray matter volume shrinkage in alcoholics. Neuropsychopharmacology. 2010;35:1120–1128. doi: 10.1038/npp.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Parada M, Corral M, Mota N, Crego A, Rodríguez Holguín S, Cadaveira F. Executive functioning and alcohol binge drinking in university students. Addictive Behaviors. 2012;37:167–172. doi: 10.1016/j.addbeh.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 131.Townshend JM, Duka T. Binge Drinking, Cognitive Performance and Mood in a Population of Young Social Drinkers. Alcoholism: Clinical and Experimental Research. 2005;29:317–325. doi: 10.1097/01.alc.0000156453.05028.f5. [DOI] [PubMed] [Google Scholar]
- 132.Litten RZ, Ryan ML, Falk DE, Reilly M, Fertig JB, Koob GF. Heterogeneity of alcohol use disorder: understanding mechanisms to advance personalized treatment. Alcoholism: Clinical and Experimental Research. 2015;39:579–584. doi: 10.1111/acer.12669. [DOI] [PubMed] [Google Scholar]
- 133.Litten RZ, Falk DE, Ryan ML, Fertig JB. Discovery, development, and adoption of medications to treat alcohol use disorder: goals for the phases of medications development. Alcoholism: Clinical and Experimental Research. 2016;40:1368–1379. doi: 10.1111/acer.13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Choi KH, Wykes T, Kurtz MM. Adjunctive pharmacotherapy for cognitive deficits in schizophrenia: meta-analytical investigation of efficacy. The British Journal of Psychiatry. 2013;203:172–178. doi: 10.1192/bjp.bp.111.107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Buchanan RW, Davis M, Goff D, Green MF, Keefe RS, Leon AC, et al. A summary of the FDA-NIMH-MATRICS workshop on clinical trial design for neurocognitive drugs for schizophrenia. Schizophrenia bulletin. 2005;31:5–19. doi: 10.1093/schbul/sbi020. [DOI] [PubMed] [Google Scholar]
- 136.Mahableshwarkar AR, Zajecka J, Jacobson W, Chen Y, Keefe RS. A randomized, placebo-controlled, active-reference, double-blind, flexible-dose study of the efficacy of vortioxetine on cognitive function in major depressive disorder. Neuropsychopharmacology. 2015;40:2025–2037. doi: 10.1038/npp.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo-controlled study of vortioxetine on cognitive function in depressed adults. International Journal of Neuropsychopharmacology. 2014;17:1557–1567. doi: 10.1017/S1461145714000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Garbutt JC, Kampov-Polevoy AB, Kalka-Juhl LS, Gallop RJ. Association of the sweet-liking phenotype and craving for alcohol with the response to naltrexone treatment in alcohol dependence: a randomized clinical trial. Jama psychiatry. 2016;73:1056–1063. doi: 10.1001/jamapsychiatry.2016.2157. [DOI] [PubMed] [Google Scholar]