

Abstract
Background
Systemic lupus erythematosus (SLE) is a chronic inflammatory, multisystem autoimmune condition. Dehydroepiandrosterone (DHEA) is a naturally occurring inactive steroid which may possess disease activity modifying properties as well as the ability to reduce flares and steroid requirements.
Objectives
To assess the effectiveness and safety of dehydroepiandrosterone compared to placebo in the treatment of people with systemic lupus erythematosus.
Search methods
We searched The Cochrane Library (Issue 2, 2006), MEDLINE, Pub Med, EMBASE, Science Citation Index and ISI Proceedings as well as searching web sites of Genelabs, FDA and EMEA. (Searches undertaken in June 2006 unless otherwise specified).
Selection criteria
We included randomised controlled trials of at least three months duration comparing DHEA to a placebo in people with SLE.
Data collection and analysis
Two review authors assessed quality and extracted data.
Main results
From the seven RCTs identified (842 participants) to date there is 'gold' ranking evidence (www.cochranemsk.org) that DHEA:
had little clinical effect on disease activity in those with mild/moderate disease (measured by SLEDAI or SLAM) but one study demonstrated evidence of stabilisation or improvement in 8.3% more patients than those treated with placebo; had a modest but clinically significant improvement in health related quality of life measured by Patient Global Assessment, estimated as 11.5% (11.5 mm on a 100 mm scale) by meta‐analysis; resulted in a greater number of patients experiencing adverse events, particularly androgenic effects such as acne where patients risk was doubled when compared to placebo (RR 2.2; 95% CI 1.65 to 2.83)
Authors' conclusions
Studying effectiveness of DHEA for SLE is difficult, reflecting the problems of studying any treatment for a disease as complex as SLE. From the seven RCTs to date, there was evidence that DHEA had a modest but clinically significant impact on health related quality of life in the short term. Impact on disease activity was inconsistent, with DHEA showing no benefit over placebo in terms of change in SLEDAI in all but one of the 6 studies reporting this outcome. Long term outcomes and safety remain unstudied.
Plain language summary
Dehydroepiandrosterone (DHEA) for lupus erythematosus
This summary of a Cochrane review presents what we know from research about the effect of dehydroepiandrosterone (DHEA) for lupus. The review shows that:
DHEA probably leads to little or no difference in disease activity in people with mild to moderate disease, but probably slightly improves overall well‐being. These results are based on moderate quality evidence. DHEA may improve disease activity in people with severe or active lupus, but this result is based on low quality evidence so there is not enough evidence to be certain. It is not known whether DHEA reduces the damage that lupus causes to organs as damage was not measured in the studies. Possible side effects may include acne, excessive hair growth, and menstrual changes. But we often do not have precise information about side effects and complications. It is not known whether DHEA causes long term side effects such as heart problems or cancer. This is because there were not many people in the studies and the longest study was only 1 year long.
What is Systemic lupus erythematosus (SLE) and DHEA (dehydroepiandrosterone)?
Systemic lupus erythematosus (SLE) or simply 'lupus' is a group of diseases in which the body's immune system attacks the body. It can affect any organ system involving connective tissue, including the skin, kidneys, eyes, lungs, heart, muscles and bones, nervous system, and gastrointestinal system. The symptoms can range from mild to life‐threatening. Lupus occurs mainly in young women, but also in men and children. DHEA (dehydroepiandrosterone) is a hormone in the body. People with lupus tend to have lower levels of DHEA, so taking DHEA supplements in pill form may help control the immune system. DHEA might also cut the need for corticosteroid treatment which means less side effects from corticosteroids.
What are the effects of DHEA (dehydroepiandrosterone)?
Disease activity (flares or changes in symptoms): We can not be sure that there is actually a difference in disease activity when taking DHEA. It is possible that these results are by chance. Overall well‐being: Improves by 12 more points on a scale of 0 to 100 Less organ damage: We are not sure whether taking DHEA could reduce organ damage because it was not measured in any of the studies. Side effects and complications: 20 out of 100 more people with mild to moderate lupus will have acne with DHEA than with no treatment.
Background
Systemic lupus erythematosus (SLE) is a chronic inflammatory, multisystem autoimmune condition mainly affecting women of childbearing age, but also men and children. The prevalence of SLE in the UK has been estimated to be 29 per 100,000 (approximately 12,000 people affected across England & Wales) (Johnson 1995), but the prevalence in certain patient groups is much higher. It has been estimated that as many as 1 in 250 black women in the USA and West Indies may have SLE, with 1 in 1000 Chinese women, and 1 in 4200 Caucasian women in New Zealand (Hart 1983; McCarty 1995; Mok 2003). Although the prevalence of SLE is high in an Afro‐Caribbean population in the USA and UK, the prevalence of lupus is low in most African countries (Nived 1997). The incidence of clinical findings in SLE also varies with ethnicity. Chinese patients have a higher incidence of renal involvement and a lower incidence of arthritis (Lee 1993) There is also evidence that there may be a substantial number of people with undiagnosed SLE in the community (Johnson 1996). SLE causes substantial morbidity although 10 year survival has improved from less than 50% in the 1950s to 90% (Ruiz‐Irastorza 2001).
There is no cure for SLE and a variety of treatments are used to manage the disease. Four main categories of systemic treatments are used in conjunction with topical treatments, physiotherapy, health promotion interventions: NSAIDs, anti‐malarials, corticosteroids, or immunosuppressants. It is important to consider that, as there is no cure, the duration of therapy may be prolonged and that toxicity should thus be minimised. NSAIDs help the arthralgia that is common in SLE, but risk GI toxicity (Singh 1999) and renal dysfunction (Whelton 2000), as seen in any population. Anti‐malarial therapy has been shown to be effective in SLE without major organ involvement (CHSG 1991). It is generally a safe and well tolerated treatment but does have a risk of retinal toxicity in 1 in 1800 cases (Silman 1997).
Corticosteroid therapy is indicated in patients with SLE when treatment with NSAIDs and hydroxychloroquine have proven inadequate in controlling the symptoms and signs of SLE. Doses around 30 mg prednisolone per day can be used to control resistant arthritis or serositis. High dose corticosteroid therapy, with doses starting at 60 mg prednisolone per day, can be used to treat the more serious aspects of SLE, such as neurological involvement, significant nephritis and haematological involvement in the form of haemolytic anaemia or thrombocytopenia. Whilst corticosteroid therapy can provide rapid improvement in patients' symptoms and signs, the dangers of long term therapy, including increased vascular risk due to hypertension and alteration to lipid profiles, increased incidence of diabetes, infection, and osteoporosis, are well established. Side effects such as weight gain, abdominal striae, acne, and Cushingoid facies can cause considerable concern and anxiety for patients. Using the lowest effective dose of corticosteroid for the shortest duration limits the potential harm. The use of bisphosphonate therapy with appropriate intake of calcium and Vitamin D has been shown to reduce the risk of corticosteroid induced osteoporosis but the other adverse events are more difficult to prevent (Homik 2004).
Immunosuppressant therapies are indicated for severe renal disease, and where disease remains active despite steroid therapy. Cyclophosphamide has been shown to be effective in severe renal disease (Boumpas 1992), but there is concern about toxicity with an increased risk of neoplasia and problems with infertility. Azathioprine may be used as a "steroid sparer" and is often seen as a less toxic option to cyclophosphamide. It does, however, have potentially serious side effects and is toxic to both the liver and bone marrow. Methotrexate has been used in a similar fashion to azathioprine but has a similar risk of harm. More recently drugs such as cyclosporin and mycophenolate mofetil have been used with some success mainly in renal lupus. Cyclosporin has been shown to improve disease control and facilitate steroid dose reduction (Caccavo 1997). More recently specific anti B cell agents, which have been used in the treatment of lymphoma, have been found to be useful in the treatment of resistant SLE. Mycophenolate Mofetil has also shown promise in the treatment of renal lupus (Kingdon 2001).
Special consideration must be given to the long term risks of corticosteroid therapy and to cytotoxic therapies, which may influence future fertility. New treatments are likely to be added to the arsenal of therapeutic options rather than replacing existing therapies and used as part of a complex care package.
Dehydroepiandrosterone (DHEA) is an inactive steroid that naturally occurs in adrenal glands, testes, ovaries and its metabolite is major circulating adrenal steroid. DHEA is differentially metabolised into a variety of potent andro/estro‐steriones in peripheral tissue including inflammatory cells. Androgens and DHEA are reduced (by ˜50%) in women with SLE, especially those with active disease, and are further reduced by corticosteroid administration. There is evidence that DHEA has an immunomodulatory effect as well as an androgenic role and both may potentially have benefits in people with SLE. In vitro ‐ DHEA reduces circulating inflammatory 'drivers' such as interleukin‐6 and up regulates interleukin‐2. In animal models, DHEA delays formation of double‐stranded DNA antibodies that characterise SLE and has been shown to improve survival (Derksen 1998; Genelabs 2001).
There is some trial evidence to suggest that DHEA may reduce disease activity, and flares. In addition, there may be a role in reducing the need for corticosteroids, and thereby reducing steroid associated osteoporosis (Genelabs 2001). DHEA may potentially have a place in therapy as an alternative to anti‐malarials, as a steroid sparing agent alone or in conjunction with other agents i.e. azathioprine, and as an adjunct to corticosteroids to reduce dose and control disease activity.
SLE is a complex condition variably affecting a range of organs and causing an array of symptoms. It is further complicated by a pattern of episodic exacerbations. Clinical trials of treatments for SLE have, therefore, been hindered by identifying effective outcomes measures that reflect disease state and patients symptoms.
The aim of this review is to summarise the evidence of effectiveness and safety of DHEA in the treatment of people with SLE.
Objectives
To assess the benefit and safety of dehydroepiandrosterone compared to placebo in the treatment of people with systemic lupus erythematosus.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials were considered eligible for the review of effectiveness. In addition, we will include observational studies and case series for assessing safety of DHEA in subsequent updates of this review.
Types of participants
Patients of any age, race, and gender who fulfilled the criteria specified by the authors for the diagnosis of systemic lupus erythematosus were included.
Types of interventions
Oral DHEA (of any dose regimen or preparation) compared with placebo therapy and given for a minimum of three months for the treatment of SLE.
Types of outcome measures
The nature of SLE, with relapsing, remitting pattern and multi organ involvement, make outcome measures complex. Biological markers for SLE do not generally correlate well with physician or patient perceived clinical outcomes. The following domains have been recommended by the outcome measures in rheumatology clinical trials (OMERACT) consensus process for assessing outcomes in clinical trials in SLE (Strand 2004) and were therefore sought as primary outcomes:
Primary outcomes * Disease Activity: Six disease activity indices have been validated in SLE in observational studies but were not designed specifically for measuring outcomes in a clinical trial setting. Ideally, disease activity would be reported as change over time, to reflect the relapsing, remitting course of SLE. We included trials with any of the validated measures of disease activity (British Isles Lupus Assessment Group scale [BILAG], SLE disease activity index [SLEDAI], SLE activity measure [SLAM], European Consensus Lupus Activity Score [ECLAM], National Institute for Health SLE Index [SIS] and Lupus Activity Index [LAI]). (Strand 1999b). * Damage: An index of irreversible end organ damage such as the American College of Rheumatology/ Systemic Lupus International Cooperating Clinics (SLICC) damage index * Health related quality of life (HRQoL): measuring the impact of the disease as perceived by the patients (Medical Outcomes Survey [SF‐20], health assessment questionnaire [HAQ] disability score, SF‐36, etc) * Adverse events
Secondary outcomes * Specific markers of organ damage: end stage renal disease * Disease flares: restricted to "major flares" defined as initiation of high dose glucocorticoid therapy, initiation or increase in immunosuppressive therapy, hospitalisation or death. * Fatigue: Krupp fatigue severity score, visual analogue scores * Steroid requirements * Steroid complications including osteoporosis
Secondary outcomes were reported in this review if recorded by the investigators, but were not included in any meta‐analysis for two reasons ‐ some of the secondary outcomes are included as elements of some of the disease activity scores; there is little consensus about the appropriateness of some of these outcomes as measures of clinical outcome in randomised controlled trials. (Strand 2004; Strand 1999b)
We sought to measure outcomes in the short term (3 to 6 months), medium term (6 to 12 months) and long term (more than 12 months).
Search methods for identification of studies
Electronic searches We searched the following databases: * The Cochrane Library ‐ all sections including the Cochrane Central Register of Controlled Trials (Issue 2, 2006) * MEDLINE (1966 to June Week 2 2006) * PubMed (limited to records added in the 60 days previous to 27 June 2006) * EMBASE (1980 to 2006 Week 25) * Science Citation Index (1980 to June 2006) * ISI Proceedings (1990 to June 2006)
Ongoing trials database * Current controlled trials (searched 27 June 2006)
Search strategy We ran the following search strategy in MEDLINE and adapted as appropriate for the other databases: 1. exp Dehydroepiandrosterone/ 2. (prasterone or dehydroepiandrosterone or DHEA or GL701 or Aslera or prestara or anastar).tw. 3. 1 or 2 4. exp Lupus Erythematosus, Systemic/ 5. (lupus or sle).tw. 6. 4 or 5 7. 3 and 6
There were no language restrictions on searching. All results were downloaded and manually screened for RCTs.
Handsearches We examined reference lists of the relevant trials and reviews identified for additional studies.
Additional searches * The web site of Genelabs (http://www.genelabs.com) * The web sites of the FDA and EMEA * The web site of EULAR http://www.eular.org/ for meeting abstracts from 2002 to 2006. * ADIS R&D Insight (27 June 2006).
Data collection and analysis
Trial selection Two observers (CB, PR) independently reviewed titles, abstracts and key words of all the records retrieved in the search. Full articles were then retrieved for further assessment when the information given suggested that the study: 1. included patients with SLE; 2. compared DHEA to placebo; 3. assessed one or more of the defined outcome measures; 4. met the defined criteria for trial inclusion.
Full articles were retrieved for clarification when there was doubt about eligibility. Inter‐rater agreement was assessed using Cohen's kappa to be 1. Had differences of opinion existed, they would have been resolved by discussion with a third party (DC). If resolving any disagreement had not been possible, the article would have been added to those 'awaiting assessment' and the authors contacted for clarification. If no clarification was provided, the review group editorial base would be consulted. No papers underwent this process.
Quality assessment The quality of reporting of each trial was based largely on the quality criteria specified by Schulz and Jadad (Schulz 1995; Jadad 1996) and as described in the manual of the Centre for Reviews and Dissemination (CRD) for randomised controlled trials (Khan 2000).
In particular the following factors were assessed: 1. Minimisation of selection bias a) was the randomisation procedure adequate? b) was the allocation concealment adequate?
2. Minimisation of attrition bias a) were withdrawals and dropouts completely described? b) was analysis by intention‐to‐treat?
3. Minimisation of detection bias a) were outcome assessors blind to the intervention?
Based on these criteria, studies were broadly subdivided into the following three categories (see Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2006): A ‐ all quality criteria met: low risk of bias. B ‐ one or more of the quality criteria only partly met: moderate risk of bias. C ‐ one or more criteria not met: high risk of bias.
This classification would have been used in the sensitivity analysis if appropriate. Two review authors independently assessed each trial. Inter‐rater agreement was calculated using Cohen's kappa to be 1. Had disagreement occurred, the rest of the group would have been consulted and a judgement made based on consensus.
Data extraction Two review authors, using a predefined data extraction form, undertook data extraction independently. Differences were resolved by discussion, with reference back to the original paper, and if necessary, a third opinion was sought.
Extraction included the following information: 1. General information ‐ author and year, country, setting, published/unpublished, language of publication, source of funding, abstract / full article, duplicate publications. 2. Trial characteristics ‐ design, duration, quality by method and security of randomisation, blinding, allocation concealment. 3. Participants ‐ age, gender, race, selection method, exclusions; duration of SLE, initial composite scores, baseline similarity of groups (including co‐morbidity), assessment of compliance, other therapies. 4. Interventions ‐ type of DHEA, dose of DHEA, placebos 5. Results ‐ comparability at baseline, losses/drop‐outs, mortality and morbidity outcomes, adverse effects, patient preference, quality of life, study duration, as observed and by intention to treat.
Data analysis Data were summarised statistically using meta‐analysis if they were available, sufficiently similar, and of sufficient quality. Dichotomous data were expressed as relative risks (RR). Continuous data were expressed as mean differences (MD) and an overall MD calculated. Overall results were calculated based on the fixed‐effect model. Heterogeneity was tested for using the Z score and the chi‐squared statistic with significance being set at P < 0.1. The I2 statistic was used to estimate the proportion of the total variation in study estimates that could be explained by heterogeneity. If significant heterogeneity was found, it was considered to be unreasonable to assume that there was one 'true' effect underlying the data, that was constant across different populations, and therefore no summary statistic was calculated.
There were insufficient studies reporting data in a way that could be summarised to enable meaningful exploration of causes of heterogeneity. In the future, if more studies are published, we will assess possible sources of heterogeneity by subgroup and sensitivity analyses as described below. Had there been sufficient studies reporting data consistently, a funnel plot would have been undertaken to assess the effect of small studies on the estimates.
Subgroup analysis We have undertaken subgroup analyses based on baseline disease severity. Only one small trial has been published in people with severe SLE. We therefore reported the findings for mild to moderate and severe separately.
We planned to perform the following additional subgroup analysis in order to explore effect size differences, had the data allowed. 1. Different types of DHEA analogues. 2. Dose. 3. Age. 4. Gender. 5. Race.
Sensitivity analyses Had it been appropriate, the following sensitivity analyses would have been performed in order to explore the influence on effect size. 1. Repeating the analysis excluding studies published in abstract form only. 2. Repeating the analysis taking account of study quality, as specified above. 3. Repeating the analysis excluding any very long or large studies to establish how much they dominated the results. 4. Repeating the analysis excluding studies using the following filters: language of publication, source of funding (industry versus other), country if appropriate.
Clinical relevance tables Clinical relevance tables were compiled under additional tables to improve the readability of the review. For any primary outcomes where a statistically significant difference between the treatment group and control group had been demonstrated and the outcome was dichotomous, like adverse events, the number needed to treat was calculated from the control group event rate and the relative risk using the Visual Rx NNT calculator (Cates 2003). For continuous primary outcomes, tables have also been presented under additional tables. When statistically significant treatment effects were demonstrated for any of the primary outcomes then the absolute benefit was calculated as the improvement in the intervention group minus the improvement in the control group, in the original units. Similarly relative difference in the change from baseline was calculated as the absolute benefit divided by the baseline mean of the control group. NNT was calculated using the Wells calculator software available at the CMSG editorial office. The minimal clinically important difference (MCID) for each outcome was determined for input into the calculator.
Grading of evidence We used the grading system described in the 2004 book Evidence‐based Rheumatology (Tugwell 2004) and recommended by the Musculoskeletal Group:
Platinum: A published systematic review that has at least two individual controlled trials each satisfying the following: * Sample sizes of at least 50 per group ‐ if these do not find a statistically significant difference, they are adequately powered for a 20% relative difference in the relevant outcome. * Blinding of patients and assessors for outcomes. * Handling of withdrawals > 80% follow up (imputations based on methods such as Last Observation Carried Forward (LOCF) are acceptable). * Concealment of treatment allocation.
Gold: At least one randomised clinical trial meeting all of the following criteria for the major outcome(s) as reported: * Sample sizes of at least 50 per group ‐ if these do not find a statistically significant difference, they are adequately powered for a 20% relative difference in the relevant outcome. * Blinding of patients and assessors for outcomes. * Handling of withdrawals > 80% follow up (imputations based on methods such as LOCF are acceptable). * Concealment of treatment allocation.
Silver: A randomised trial that does not meet the above criteria. Silver ranking would also include evidence from at least one study of non‐randomised cohorts that did and did not receive the therapy, or evidence from at least one high quality case‐control study. A randomised trial with a 'head‐to‐head' comparison of agents would be considered silver level ranking unless a reference were provided to a comparison of one of the agents to placebo showing at least a 20% relative difference.
Bronze: The bronze ranking is given to evidence if at least one high quality case series without controls (including simple before/after studies in which patients act as their own control) or if the conclusion is derived from expert opinion based on clinical experience without reference to any of the foregoing (for example, argument from physiology, bench research or first principles).
Results
Description of studies
The searches retrieved a total of 480 references. After the removal of duplicates there were 325 unique references. These contained seven RCTs, published in eight full papers (Petri 2004 comprised two separate publications), which met our inclusion criteria.
Excluded studies Three full publications (van Vollenhoven 1994; Barry 1998; Chang 2004) and five abstracts (van Vollenhoven 1992; van Vollenhoven 1993; vanVollenhoven 1994b; van Vollenhoven 1996; van Vollenhoven 2001) were excluded from this review.
All apart from Chang 2004 and van Vollenhoven 2001 were case series and excluded on the basis of study design.
Chang 2004 was conducted as a sub‐study of Chang 2002 (a multi centre randomised placebo controlled trial over 24 weeks) and evaluated the role of cytokines in treatment of SLE with DHEA. It was excluded because this was not an outcome measures included in this review.
van Vollenhoven 2001 reported a double‐blinded placebo controlled trial assessing the effect of DHEA on cognitive function in participants with mild to moderate SLE over a one month treatment period. It was excluded from this review because it was less than three months in duration.
Included Studies We identified seven studies for inclusion in this review. One of the studies had been reported in more than one full publication (Van Vollenhoven 1995) and several had been reported as a series of abstracts prior to full publication.
Study design All seven included studies were RCTs of at least three months duration. All had been published in full. The shortest of the trials lasted three months (Van Vollenhoven 1995), three lasted for six months (Van Vollenhoven 1999; Chang 2002; Nordmark 2005), one for up to nine months (Petri 2002) and the longest trials studied people receiving DHEA for up to 12 months (Petri 2004; Hartkamp 2004). Two studies (Chang 2002; Petri 2004) included a six week period prior to participation in the study during which participants must have been stable on all existing therapy, and one study required an eight week stable period (Nordmark 2005). One study (Hartkamp 2004) required participants to be stable without steroids for at least six months. No specific justification was presented by the researchers for the 'run in' periods but in all cases, the study protocol specified that no change in other medication (other than the study drug) could occur during the trial. A further study (Petri 2002) required at least six weeks of stable prednisolone therapy, and four weeks stable on any other therapy. In this case the aim of the study was to reduce the prednisolone dose. Participants There were a total of 842 participants, of which 450 were on DHEA, but after the authors had excluded selected drop outs (two dropped out from Van Vollenhoven 1995; four dropped out from Nordmark 2005) 836 people were included in the analysis. Reflecting the epidemiology of SLE, most of the trials included only women. One study (Van Vollenhoven 1999) did not exclude male participants, but only 3 of the 19 participants were male. Caucasians were the most commonly represented racial group, although one trial included only Chinese women (Chang 2002). Where reported, the median age of trial participants was approximately 30 to 47 years. One study did not report the age of the study populations (Chang 2002).
Two studies restricted participation to people with active SLE as defined by a rheumatologist, using two disease activity scales (SLEDAI > 2 and SLAM >= 7 plus steroids use of less than 10 mg per day) (Chang 2002; Petri 2004). Van Vollenhoven 1995 defined participants as having "mild to moderate" disease but did not define this further. A further study restricted participants to those with SLE managed on less than 10 mg prednisolone (Hartkamp 2004). One study (Van Vollenhoven 1999) was restricted to people with severe SLE based on meeting the ACR criteria for severe renal, haematological or serosal disease. Petri 2002 only included people receiving 10 to 30mg of prednisolone per day. Nordmark 2005 limited the study population to those receiving at least 5mg of prednisolone daily.
Those with severe disease have been reported separately.
Interventions DHEA was administered as a once daily oral dose in all but one (Nordmark 2005) of the trials. In one study, participants receiving active treatment were given 100 mg per day (Van Vollenhoven 1995). In four studies the active dose was 200 mg per day (Van Vollenhoven 1999; Chang 2002; Hartkamp 2004; Petri 2004). Petri 2002 included two active treatment arms with 100 and 200 mg daily doses in each (Petri 2002). Finally Nordmark 2005 administered low doses of DHEA (10 to 15 mg) twice daily to participants. All the trials included a placebo arm.
In total 450 people were exposed to DHEA. Seventy seven participants were treated with DHEA 100 mg daily. Three hundred and seventy three were treated with DHEA 200 mg daily. Three hundred and eighty six participants received a placebo.
Outcome measures Primary outcomes There is no single accepted measure of SLE activity and this was reflected in the range of measures reported in the seven included studies.
Disease Activity SLEDAI was reported in six of the seven studies (Petri 2002 did not) and SLAM was reported in Van Vollenhoven 1999; Chang 2002; Petri 2004. Petri 2002 states that SLEDAI was measured but does not report data because "according to the trial design, a reduction in the dose of prednisolone at protocol specified visits was mandatory when a participant's SLEDAI score was stable or had improved. SLEDAI and other secondary outcome measures would not be expected to improve". Three studies (Van Vollenhoven 1999; Petri 2002; Petri 2004) used "responder" measures as the primary outcome. In one (Petri 2004) a composite score was used based on improvement or stabilization of a number of predefined measures of disease activity. Petri 2002 defined responders to be anyone who achieved a reduction in prednisolone to 7.5 mg or less per day for two consecutive months. Van Vollenhoven 1999 defined responders as those meeting absolute predefined improvements in the main organ affected by their SLE.
The number of participants experiencing 'flares' of disease and the 'time to flare' were also reported as markers of disease activity (Van Vollenhoven 1995; Chang 2002; Petri 2004)
Damage Only one of the studies used an index of irreversible end organ damage such as the American College of Rheumatology/ Systemic Lupus International Cooperating Clinics (SLICC) damage index (Nordmark 2005) reporting that it was unchanged in both the treatment and placebo groups but with no further details (one stated that it was measured but did not report any results Petri 2002). Some reported individual end organ damage (Van Vollenhoven 1999 reported renal damage) or included assessment of organ damage as part of their composite scoring index (Van Vollenhoven 1999; Petri 2004).
Health related quality of life Only one of the studies reported health related quality of life using methods other than patient global visual analogue scales (Nordmark 2005). The authors reported a number of tools including Short Form‐36, a Hopkins Symptom Checklist (HSCL) and the psychological General Well‐Being index as well as a Swedish version of the McCoy Sex Scale Questionnaire. Four of the other studies reported patient global scores (Van Vollenhoven 1995; Van Vollenhoven 1999; Chang 2002; Petri 2004). The Patient global visual analogue scale is a simple generic tool allowing participants to indicate their perception of their health status on a 100 mm bar where 100 represents optimal and zero represents the worst possible. There are a number of disease specific questionnaires for use in SLE, including the SLEQoL and LupusQoL. None of these scales were reported. One study stated that the primary endpoint was well‐being and fatigue, but did not report any results for either of these endpoints (Hartkamp 2004).
Secondary Outcomes The ability to reduce concurrent corticosteroid dose was assessed as the primary outcome in one study in the form of a "responder" definition (Petri 2002) but was also reported in two other studies (Van Vollenhoven 1995; Van Vollenhoven 1999). Changes in bone mineral density was measured in three studies (Van Vollenhoven 1999; Hartkamp 2004; Nordmark 2005) and for a subgroup of Petri 2004. Different summary measures were reported for each study.
Krupp fatigue severity score was reported in Petri 2004.
The 'Characteristics of included studies' table details the outcome measures included by each study.
Risk of bias in included studies
Details of the quality assessment is summarised in Table 1.
1. Summary of quality assessment of included studies.
| Study id | Assignment | Alloc concealment | Baseline | ITT | Dropouts | Outcome Blind |
| Chang 2002 | Yes | Probably Adequate | No detail presented | Yes | Yes (7/120) | Yes |
| Hartkamp 2004 | Unclear | Adequate | No (postmenopausal status higher in active arm; oestrogen use lower in active arm) | No (2 declined final dexa scan excluded | Yes (2/60) | Yes |
| Nordmark 2005 | Unclear | Unclear | Similar (DHEA treatment group had slightly more active disease | Unclear | Yes (3/41) | Yes |
| Petri 2002 | Unclear | Probably Adequate | Yes | Yes | No (49/191) | Yes |
| Petri 2004 | yes | Adequate | yes (although Anti dsDNA higher in active treatment group) | Yes | Yes (115/381) | Yes |
| Van Vollenhoven 1995 | Unclear | Unclear | Yes | No | Yes (2/28) | Yes |
| Van Vollenhoven 1999 | Yes | Probably Adequate | No Males in placebo group only, organs effected differed.Some differences in baseline activity scores with DHEA group tending to be higher (none statistically significant) | No | Yes (2/21) | Yes |
| KEY | Assignment random | Allocation concealed | Baseline characteristics similar | Intention to treat analysis | drop outs described | Outcomes blinded |
Only one of the trials was considered to fall into category A of the quality assessment detailed in the methods, minimising the risk of selection, attrition and detection bias (Petri 2004). Van Vollenhoven 1995, Petri 2002, and Hartkamp 2004 reported sparse (or no) details of the assignment and allocation concealment methods. Losses to follow up were poorly reported in two of the studies (Chang 2002; Petri 2002). Chang 2002 provided no information about the baseline characteristics of the study groups. Baseline characteristics in Van Vollenhoven 1999 showed some potentially important differences including the only male participants (N = 3) receiving placebo and substantial differences in the distribution of severely effected organs between treatment and placebo arms. Nordmark 2005 excluded four people who dropped out during the study period as a result of adverse events. It is not clear when these people withdrew from the study, but all four appear to have been in the active treatment group. We have retained these four people in the analysis of participants. Van Vollenhoven 1995 also excluded two people, one from each group. In this study withdrawal occurred before any treatment had been administered and therefore the authors decision to excluded them from analysis has been reflected in our analysis of participants.
In addition, two of the studies were very small, containing less than 30 participants (Van Vollenhoven 1995; Van Vollenhoven 1999).
Petri 2002 and Petri 2004 performed well against the quality criteria set out in the method. However, Petri 2002 undertook post hoc subgroup analysis, after unblinding, because initial analysis identified large number of participants experiencing a positive primary outcome (which included "stable disease") in both the treated and untreated groups. The researchers found that response correlated with SLEDAI score, with higher scores experiencing poorer responses in the placebo group. They presented results by subgroups of SLEDAI score at baseline. Petri 2004 was complicated by a change in 'entry' requirements during the study period. The change, to include separate analysis for those with SLEDAI > 2, was added as a result of the findings of an earlier study (Petri 2002) which reported substantial differences in response among those with higher SLEDAI. The change to protocol was undertaken prior to the unblinding of the study. The authors, therefore, present two analyses for their primary outcome ‐ a composite measure; for all randomised participants, and for those meeting a stricter definition of 'active SLE' defined by SLEDAI > 2. For other outcome measures ‐ only those with SLEDAI > 2 were reported.
Effects of interventions
Primary outcomes Disease activity Of the six widely used disease activity measures only two were reported by any of the studies, the SLEDAI and the SLAM. The SLEDAI and SLAM are both global scoring systems which aim to provide an overall assessment of disease activity rather than being assessments of individual organ systems.
SLEDAI The SLEDAI measures disease activity in the preceding ten days. It contains 24 weighted parameters and scores can range from zero to 105. Although it is possible to score 105 this is rarely seen in clinical practice, and a score of 20 represents very high disease activity. A SLEDAI score of 11 to 19 represents high disease activity, whilst 6 to 10 is moderate activity, and 1 to 5 is mild. Zero represents no disease activity.
Six of the studies reported disease activity using SLEDAI (Van Vollenhoven 1995; Van Vollenhoven 1999; Chang 2002; Hartkamp 2004; Petri 2004; Nordmark 2005). Four studies (Van Vollenhoven 1995; Van Vollenhoven 1999; Chang 2002; Hartkamp 2004) reported scores and one study (Petri 2004) reported, for the subgroup of participants who had active disease at baseline (defined as SLEDAI > 2), the percentage of participants who remained 'stable or improved'. One (Nordmark 2005) reported that there was no change in SLEDAI from baseline but gave no further details.
Mild to Moderate SLE None of the studies reported statistically significant change in SLEDAI score from base line when comparing the treatment groups to the placebo group on an intention to treat basis. Three studies reported small changes in SLEDAI in the placebo and treatment arms (mean difference in change from baseline of less than 1 point on the scale) (Chang 2002; Hartkamp 2004; Nordmark 2005).
Meta‐analysis was possible for two of the studies (N = 148) (Van Vollenhoven 1995; Chang 2002) and supported no statistically significant mean difference in SLEDAI score among those treated with DHEA versus placebo (‐0.6; 95% CI ‐2.12 to 0.89). Ranking of evidence: Gold based on the meta‐analysis of two studies (N = 148 with one RCT containing > 50 participants in each treatment group and the larger study (Chang 2002) being of good methodological quality). Three of the studies that could not be included in the meta‐analysis reported findings in keeping with the meta‐analysis result.
Petri 2004 reported 90.5% of participants experiencing a stabilisation or improvement in the SLEDAI score on treatment versus 82.2% of participants stabilising or improving without treatment (P = 0.04) if the analysis was restricted to those with active disease (SLEDAI > 2). This did not give any measure of the scale of change in SLEDAI detected in those who improved or those who deteriorated. Using the more lenient requirement of "stabilisation or improvement" in condition, Petri 2004 reported that a statistically significantly greater proportion of participants treated with DHEA met the criteria compared to placebo (8.3% stabilised or improved P = 0.04). This was gold ranking evidence.
Severe SLE One small study, in people with severe disease, reported a mean improvement in SLEDAI score of 10.3 compared to an improvement of 3.9 in the placebo group (Van Vollenhoven 1999). (Number needed to treat (NNT)= 3) This was of borderline statistical significance, however, only 19 people participated in this study. Ranking of evidence: silver
SLAM The SLAM measures disease activity in the preceding month. The original index was published in 1989 and has been subsequently revised to the SLAM‐R. It measures 30 variables in 11 organ systems and 8 laboratory features. The minimum score is zero and the maximum is 84. A score of 7 is felt to indicate disease activity that would warrant modification of treatment in 50% of participants.
SLAM was reported in three studies (Petri 2004; Chang 2002; Van Vollenhoven 1999). Two of the studies reported scores (Van Vollenhoven 1999; Chang 2002) and one study (Petri 2004) percentage of participants who 'stabilised or improved' but only in a subgroup of those with active disease (SLEDAI > 2). None of the studies demonstrated a statistically significant change in the SLAM between the treatment and placebo groups. Ranking of evidence: gold*
*Gold ranking evidence based on the meta‐analysis of two studies (N =148 with one RCT containing > 50 participants in each treatment group and the larger study (Chang 2002) being of good methodological quality). Petri 2004 reported findings consistent with no statistically significant difference in SLAM scores for those treated with DHEA as compared to placebo. Table 2; Table 3; Fig 01,01 summarises the results.
2. Summary of Disease activity: SLEDAI (0‐105) (individual studies).
| Study | no. participants | placebo (SEM) | DHEA (SEM) | change vs placebo | ||
| Mild/Moderate | ||||||
| Chang 2002* | Placebo: 59; DHEA 61 | ‐1.4 (4.6) | ‐1.2 (5.4) | p=0.7 | ||
| Hartkamp 2004 | Placebo: 30; DHEA 30 | +0.3 (na) | +0.43 (na) | p=0.79 | ||
| Nordmark 2005 | Placebo: 17 DHEA 20 | reported as "no significant change" | ||||
| Petri 2002 | Placebo 64; DHEA 100 63; DHEA 200 64 | measured but not reported for all participants | ||||
| Petri 2004 | Placebo: 192 (146 reported) DHEA 189 (147 reported) | 17.8% deteriorated | 9.5% deteriorated | p=0.04 not reported for all participants ‐ only those with "active disease" | ||
| van Vollenhoven 1995* | Placebo: 14 DHEA 14 | +0.79 (0.75) | ‐1.71 (1.18) | p=0.09 | ||
| *Summarised in Meta‐analysis | ||||||
| Severe | ||||||
| van Vollenhoven 1999* | Placebo: 10 DHEA 9 | ‐3.9 (1.4) | ‐10.3 (3.1) | p=0.07 | ||
| *Summarised in Meta‐analysis |
3. Summary of Disease activity: SLAM (0‐84) (individual studies).
| Study | No. participants | placebo (SEM) | DHEA (SEM) | change vs. placebo | |||
| Mild/Moderate Disease at baseline | |||||||
| Chang 2002* | Placebo: 59; DHEA 61 | ‐2.0 (0.49) | ‐2.6 (0.44) | p=0.355 | |||
| Petri 2004 | Placebo: 192 (146 reported) DHEA 189 (147 reported) | 10.3% deteriorated | 6.8% deteriorated | p= 0.29 Not reported for all participants ‐ only those with "active disease" | |||
| Severe disease at baseline | |||||||
| van Vollenhoven 1999 | Placebo: 10 DHEA: 9 | ‐2.4 (2.0) | ‐5.4 (2.34) | p=0.41 |
Summary for Disease Activity : There was 'gold' level evidence and consistent findings across five studies that, in people with mild to moderate SLE, treatment with DHEA provided no benefit over placebo in terms of disease activity, measured by mean change in SLEDAI.
However, there was also 'gold' level evidence from a single study that, if only those with 'active' disease (SLEDAI > 2) were included in the analysis, 8.3% (P = 0.04) more people remain "stable or improve" on DHEA versus placebo, measured using SLEDAI .
This inconsistency in the evidence may arise because the criteria of "stabilised or improved" is considered to be a more lenient assessment than mean change in SLEDAI score. The inconsistency between findings does not seem to be explained by the restriction to only those with SLEDAI > 2 at baseline because the Chang 2002 study also included this restriction but found no evidence of improvement in SLEDAI score.
One very small trial in participants with severe SLE reported improvements in SLEDAI among those treated with DHEA that were of borderline statistical significance when compared with placebo.
There was consistent gold ranking evidence that DHEA did not provide clinical benefits when measuring disease activity using SLAM.
Organ damage As SLE is a chronic multisystem disease it is important to assess chronic organ damage. The SLICC Damage index has been endorsed by the American College of Rheumatology and is the most widely used damage index. It assesses 41 variables in 12 systems and the damage must persist continuously for six months to score. The maximum possible score is 47, but it would be uncommon in clinical practice for participants to survive with scores = 12. Only approximately 50% of SLE participants would be expected to score on the SLICC/ACR damage index.
None of the trials presented data on SLICC/ACR damage index scores.
Health Related Quality of life In any chronic disease the patient's perception of their health related quality of life is important. The most widely used survey is the generic SF36 (Short Form 36) which assesses eight domains of general health perception, physical and social functioning, limitation due to physical or emotional factors, mental health, vitality and pain. Lower scores represent poorer health status. Only one study reported SF36, while four reported Patient or Physician Global Assessment using a 0 to 100mm visual analogue scale. A clinically relevant change in Patient or Physician Global Assessment has been reported to be +/‐10mm.
SF‐36 Mild to Moderate SLE Only Nordmark 2005 reported SF36, finding improvements in SF36 among those treated with low dose DHEA, when compared to those getting placebo, in the functional domains of role limitation due to physical factors and to emotional factors. This was of statistical significance in the domain of role limitation due to emotional factors (+ 23.3 versus ‐14.6). Ranking of evidence: silver (small study of < 50 participants in each group)
Patient Global Assessment Four of the studies reported the generic Patient Global Assessment (Van Vollenhoven 1995; Van Vollenhoven 1999; Chang 2002; Petri 2004). Another two studies measured but did not report Patient Global Assessment (Petri 2002; Hartkamp 2004).
Mild to Moderate SLE Both Chang 2002 and Van Vollenhoven 1995 reported statistically significant improvements in the Patient Global Assessment in the treatment group compared to deterioration in the placebo (after adjustment for other study factors).
Meta analysis was possible for these two studies and supported a statistically significant mean difference in Patient Global Assessment (11.5% reduction (reduction of 11.5 mm; 95% CI ‐19.08 to ‐3.84) on a scale of 0 to 100 mm) ] in those treated with DHEA versus those receiving placebo. (NNT to achieve this level of improvement =5) Ranking of evidence: Gold (N = 148 with one RCT containing > 50 participants in each treatment group and the larger study (Chang 2002) being of good methodological quality).
One study (Petri 2004) reported that, for the subgroup of participants with active disease (SLEDAI > 2) a statistically significant lower percentage of participants deteriorated in their Patient Global Assessment in the DHEA group (10.9%) compared to placebo (22.6%) (P = 0.007).
Severe SLE One study (Van Vollenhoven 1999) found no difference in Patient Global Assessment between treatment and placebo groups. Ranking of evidence: silver.
Table 4 and Fig 0201 summarises these findings.
4. Summary of Health Related Quality of Life: Patient Global (0‐100) (ind.studies).
| study | No. Participants | placebo (SEM) | DHEA (SEM) | change vs. placebo | ||
| Mild to Moderate disease | ||||||
| Chang 2002 | Placebo 59: DHEA 61 | +5.4 (2.56) | ‐5.5 (3.46) | p=0.005 (adjusted for treat centre and treatment, no unadjusted presented) [95% CI from crude analysis ‐76.1 to 54.3] | ||
| Hartkamp 2004 | Placebo 30: DHEA 30 | measured but not reported | ||||
| Nordmark 2005 | Placebo 17: DHEA 20 | not measured | ||||
| Petri 2002 | Placebo 64: DHEA 100mg 62: DHEA 200mg 64 | measured but not reported | ||||
| Petri 2004 | Placebo 192 (reported 146): DHEA 189 (reported 147) | 22.6% deteriorated | 10.9% deteriorated | p=0.007 Not reported for all participants, only those with "active disease" | ||
| van Vollenhoven 1995 | Placebo 14: DHEA 14 | +2.4 (7.0) | ‐11.5 (5.7) | unadjusted 0.138, P vs placebo adjusted 0.022 | ||
| *Summarised in Meta‐analysis | ||||||
| Severe Disease | ||||||
| van Vollenhoven 1999 | Placebo 10: DHEA 9 | ‐23.5 (2.4) | ‐23.7 (4.8) | p= 0.53 |
Physicians Global Score The Physicians global score was reported in three of the studies (Van Vollenhoven 1995; Van Vollenhoven 1999; Chang 2002). In none of the studies was a statistically significant improvement achieved.
Mild to moderate SLE Meta‐analysis was possible for the two studies (Van Vollenhoven 1995; Chang 2002) and supported no statistically significant mean difference in Physician Global Score (‐3.2; 95% CI ‐8.1 to 1.8) for treatment with DHEA versus placebo. Ranking of evidence: gold (N = 148 with one RCT containing > 50 participants in each treatment group and the larger study (Chang 2002) being of good methodological quality).
Hartkamp 2004 stated that the main outcome of interest in their study was wellbeing and fatigue. We can find no report of these endpoints in the published literature.
Summary of Health Related Quality of Life : There was 'gold' level and consistent evidence that treatment with DHEA did statistically significantly improve quality of life, measured by Patient Global Assessment, when compared with placebo. There was evidence of improvement in some, but not all, domains of the SF36 tool when treated with low dose DHEA. Using Physician Global Assessment, there was gold level and consistent evidence of no clinical benefit.
Adverse Events Table 5 and Figs 0301 to 0303 summarises the adverse events Adverse events were not consistently reported throughout the studies. It should be noted that two studies (Hartkamp 2004; Nordmark 2005) did not report any information about adverse events. Due to DHEA androgenic properties, we anticipated that certain adverse events might be reported (cardiovascular disease and hypertension, acne and hirsutism, cancer). These will be considered specifically below but general adverse event reporting is outlined first.
5. Adverse events.
| Adverse events | Chang 2002 | Hartkamp 2004 | Petri 2002 | Petri 2004 | van Vollenhoven 1995 | van Vollenhoven 1999 | Nordmark 1002 |
| No. Participants | DHEA:61; Placebo:59 | DHEA:30; Placebo:30 | DHEA 100mg:63; DHEA 200mg: 64; Placebo:64 | DHEA:192; Placebo: 189 | DHEA:14; Placebo:14 | DHEA:10; Placebo:10 | DHEA: 20; Placebo:17 |
| Serious AEs | DHEA: 7 (11.5); Placebo: 18 (30.5) | DHEA: 33 (17); placebo: 27 (14) | DHEA: 1; placebo: 1 | DHEA: 1; placebo: 2 | |||
| Withdrawal due to AE | DHEA 100: 4 (6); DHEA 200: 6 (9); placebo:3 (5) | DHEA: 11 (5.7); placebo:27 (14.3) | DHEA: 0; placebo: 0 | ||||
| Acne | DHEA: 59%; placebo: 29% | DHEA 100: 26 (41); DHEA 200: 26 (41); placebo:12 (19) | DHEA: 63 (33.3); placebo: 27 (14.1) | DHEA:8 (57); placebo: 1 (7) | DHEA: 6; placebo:3 | ||
| Hirsutism | DHEA 100: 7 (11); DHEA 200:5 (7.8); placebo: 3 (4.7) | DHEA: 31 (16.4); placebo: 3 (1.6) | DHEA: 2 (14); placebo:4 (28) | DHEA:4; placebo:2 | |||
| Weight Gai | DHEA: 2(14); placebo:1 (7) | ||||||
| Rash | DHEA 100: 3 (4.8); DHEA 200: 7 (11); placebo: 3 (4.7) | DHEA: 62 (32.3); placebo: 75 (39.7) | DHEA :0 ; placebo: 2 (14) | ||||
| Menarrhagia | DHEA 100:5 (7.9); DHEA 200: 5 (7.8); placebo: 3 (4.7) | DHEA: 1 (7); placebo: 2 (14) | DHEA: 8; placebo :3 | ||||
| Headache | DHEA 100: 3 (4.8); DHEA 200: 4 (6.3); placebo:1 (1.6) | DHEA: 56 (29.2); placebo:42 (22.2) | DHEA:4 ; placebo: 4 | ||||
| Abdominal Pain | DHEA 100: 3 (4.8); DHEA 200: 5 (7.8); placebo:0 | DHEA: 30 (25.6); placebo: 27 (14.3) | |||||
| Chest Pain | DHEA: 22 (10.4); placebo: 14 (7.4) | ||||||
| Arthralgia | DHEA:71 (37.0); placebo: 68 (36) | ||||||
| Asthenia | DHEA 100: 4 (6.3); DHEA 200: 3 (4.7); placebo:3 (4.7) | DHEA: 51 (26.6); Placebo45 (23.8) | |||||
| Myalgia | DHEA: 69 (35.9); placebo: 42 (22.2) | ||||||
| flu like symptoms | DHEA: 42 (21.9); placebo: 39 (20.6) | ||||||
| Stomatitis | DHEA: 44 (22.9); placebo: 28 (14.8) | ||||||
| Mood Change | DHEA: 30 (15.6); placebo:28 (14.8) | DHEA; 1 (7); placebo: 0 | DHEA:1; placebo:3 | ||||
| Alopecia | DHEA: 39 (20.3); placebo: 28 (14.8) | DHEA: 0; placebo:1 | |||||
| Fever | DHEA: 28 (14.6); placebo: 22 (11.6) | ||||||
| Perihperal vascular disease | DHEA: 20 (10.4); placebo: 19 (10.1) | ||||||
| Sinusitis | DHEA: 21 (10.4); placebo: 17 (9) | ||||||
| Insomnia | DHEA 100: 4 (6.3); DHEA 200: 3 (4.7); placebo:2 (3.1) | DHEA: 2; placebo:1 |
Withdrawal of treatment due to adverse events Two studies reported withdrawals of treatment thought to relate to adverse events (Petri 2002; Petri 2004). Petri 2004 reported statistically significantly more withdrawals due to adverse events among those treated with DHEA (14.3% versus 5.7%; P = 0.005). Most of this difference was attributed to androgenic adverse events (acne and hirsutism). Similar rates of withdrawal in active and control groups were reported in the other study (Petri 2002) with 5% withdrawal in the placebo and 6 to 9% in the DHEA groups (two different drug doses). Van Vollenhoven 1995 noted that there were no withdrawals from treatment due to adverse drug reactions.
Serious adverse events Four of the studies reported an overview of adverse events classed as 'serious' by the assessing physicians (Van Vollenhoven 1995; Van Vollenhoven 1999; Chang 2002; Petri 2004). Petri 2004 reported 14% of participants receiving DHEA as experiencing serious adverse events compared to 17% on placebo. Most of these (16/33 on placebo; 14/27 on DHEA) resulted in cessation of the study drug. No deaths were experienced in the DHEA treated group but five occurred among those receiving placebo (1 x pulmonary hypertension; 2 x suicide; 1 x sudden death; 1 x non‐Hodgkin's lymphoma). Van Vollenhoven 1999 reported only three serious adverse events: one death in the DHEA group from SLE complications shortly after randomisation; one deep venous thrombosis and one with fever associated with leucopaenia in the placebo group). In Chang 2002, 11.5% on DHEA experience serious adverse events, compared with 30.5% on placebo (P = 0.01) but this included events that were consistent with SLE disease flares. Van Vollenhoven 1995 noted that two serious adverse events had been reported, one in placebo and one receiving 200 mg DHEA. No further details were given. NB no standard definition of "serious" was reported.
Cardiovascular events None of the studies reported on the effect of DHEA on blood pressure. Petri 2004 reported chest pain among 10.4% of those receiving DHEA versus 7.4% of those on placebo, a difference that was not statistically significant and no further details were given. None of the other trials reported on chest pain or any other cardiac events.
Acne, hirsutism and menstrual change (Figures 03/01; 03/02; 03/03) Acne was the most commonly reported adverse event for DHEA occurring in between 33% and 59% of people receiving DHEA. That compares with 7 to 29% reporting acne in the placebo groups. Hirsutism, weight gain, and menstrual change where among the other androgenic side effects reported.
Mild to moderate SLE Meta‐analysis of the four studies presenting data for acne in a suitable format (Van Vollenhoven 1995; Chang 2002; Petri 2002; Petri 2004;) estimated the relative risk of acne to be 2.16 (95% CI 1.65 to 2.83) in those treated with DHEA versus those on placebo. This difference was statistically significant. For hirsutism, the studies reported heterogeneous results, varying from no statistical difference to placebo to a relative risk of 10.2 associated with treatment. As no clear definition of hirsutism was given by any of the trialists, we have not presented a summary statistic for this adverse event. No statistical difference was reported for menstrual change (Relative risk 1.2; 95% CI 0.38 to 3.77). (Number Needed to Treat to obtain this level of acne adverse events = 5) Ranking of evidence: Gold.
Severe SLE From the one small study, no statistically significant difference in acne or hirsutism was reported between the treatment groups. Menstrual change was reported more often by those treated with DHEA (Relative Risk 2.67; 95% CI 0.98 to 7.22) (Van Vollenhoven 1999). Ranking of evidence: Silver.
Cancer Only one study reported identifying cancers (Petri 2004). All three tumours occurred in the population receiving placebo. One was a fatal non‐Hodgkin's lymphoma, the other two were carcinoma of the lung and breast.
Other reported adverse events Petri 2004 provided the most comprehensive reporting of adverse events. Headache was reported in 56 of the 192 participants receiving DHEA (29.2% versus 22.2% placebo); 30 reported abdominal pain (25.6% versus 14.3%); 30 reported mood change (15.6% versus 14.8%); and 21 reported sinusitis (10.4% versus 9.0%). Musculoskeletal symptoms, rashes, fever, peripheral vascular disease and flu like symptoms were also reported among participants but are difficult to distinguish from disease activity.
Long term and rare adverse events The longest exposure to DHEA in any of the randomised trials was 12 months (Hartkamp 2004; Petri 2004) but as noted above Hartkamp 2004 did not report on adverse events. Total exposed patient experience from the included RCTs was limited to 450 people who received active therapy.
Secondary outcomes In addition to the primary outcomes above, we identified, a priori, the following secondary outcomes as of clinical relevance. These outcomes were reported inconsistently in all trials.
Specific Organ Damage Markers One study (Van Vollenhoven 1999) presented data on change in proteinuria over six months, with reductions seen in both the placebo and DHEA group and no statistically significant difference between the groups.
Flares Defining a flare in SLE is difficult as it can represent anything from major organ involvement to a change in the patient's social environment. Some investigators base the definition on change in the disease activity score whilst others require change in organ specific manifestations of the disease. Different definitions of a flare where used in each of the three studies reporting on flares making comparisons across studies impossible.
One study (Petri 2004) gave a five point criteria for defining a flare and, in addition, reported time to flare. One study (Chang 2002) modified the description of a flare from the SELENA study (FitzGerald 1999; Petri 2005) by increasing the glucocorticoid dose by 2.5 mg for at least seven days for SLE related reasons. These authors also reported time to first flare. One study (Van Vollenhoven 1995) assessed flares retrospectively based solely on whether the term "flare" was used in the patient's chart by the primary rheumatologist.
In all three studies (Van Vollenhoven 1995; Chang 2002; Petri 2004) a smaller percentage of participants flared in the DHEA group compared to the placebo group (Petri 2004 placebo 23.8% versus DHEA 29.7%, P = 0.27 (significant reduction if analysis restricted to those with active disease); 34% versus 18%, P not significant; 57% versus 21%, P = 0.053).
Fatigue Fatigue can be a disabling symptom in SLE. Whilst fatigue has been shown to influence scoring on the SF36, there are specific tools available to assess fatigue. The Krupp Fatigue Severity Score was developed to assess fatigue in participants with Multiple Sclerosis and SLE. It is a nine items with a seven‐point response with high scores representing greater perception of fatigue. One study (Petri 2004) reported the mean change in score with 14.4% of participants on placebo deteriorating compared to 10.9% of those on DHEA; this was not statistically significant (only reported for those with active disease).
Steroid Use Corticosteroid therapy has an important role in the management of SLE. Therapy may require to be initiated in response to an increase in disease activity or the dose of corticosteroid may require to be increased if the patient is already on maintenance corticosteroid therapy.
Three studies reported on corticosteroid use (Van Vollenhoven 1995; Van Vollenhoven 1999; Petri 2002). Three studies reported the mean change in corticosteroid use from baseline to final visit (Van Vollenhoven 1995; Van Vollenhoven 1999; Petri 2002) whilst one study (Petri 2002) also reported the number of days patient in the study were maintained on a dose of 7.5 mg or less of prednisone.
Change in steroid dose (Van Vollenhoven 1995; Van Vollenhoven 1999) showed a reduction in corticosteroid use in the DHEA group compared to placebo in all three studies, but none were statistically significantly different from the placebo group. In Van Vollenhoven 1999 a mean reduction of 36.2 mg was achieved in the DHEA group as compared with a reduction of 30.2 mg in the placebo group (P = 0.58). Van Vollenhoven 1995 reported a mean reduction of 3.2 mg in the DHEA group versus a 2.4 mg increase in the placebo group (P = 0.1). Petri 2002 reported a 36% reduction in mean prednisolone dose in the placebo group versus a 30% reduction in the 200 mg DHEA group (P = 0.67).
The one study (Petri 2002) which reported on the number of days participants were maintained on a dose of 7.5 mg or less of prednisone during the study period required participants to be on corticosteroid doses of between 10 to 30 mg of prednisolone for a period of 12 months prior to recruitment to meet the inclusion criteria. The study was designed with the aim of reducing steroid dose and if participants' disease was stable or improved, then steroids were reduced. The investigators found participants on placebo spent a median of 66.5 days on 7.5 mg or less of prednisolone. Participants on 100 mg of DHEA spent a median of 81 days on doses of prednisolone of 7.5 mg or less, whilst those on 200 mg of DHEA spent a median of 111.5 days on doses of 7.5 mg prednisolone or less. This finding was statistically significant for those on 200 mg DHEA versus placebo (P = 0.07). This study noted a high proportion of those with low SLEDAI scores managed to maintain prednisolone doses of less than 7.5 mg per day regardless of treatment.
Post hoc subgroup analysis of those with SLEDAI scores greater than 2 at baseline reported similar findings with a median of 28 days on a dose of 7.5 mg or less prednisolone for those on placebo (N = 45) and 110 days for those on 200 mg DHEA (N = 45) (P = 0.013).
Steroid Complications Prolonged use of corticosteroids is clearly associated with corticosteroid related side effects such as osteoporosis, hypertension and diabetes mellitus. As participants are now surviving far longer with their disease the toxicity of therapies is becoming increasingly relevant. Awareness of this was one of the driving forces behind the development of the SLICC/ACR damage index. One of the points on the SLICC/ACR index is osteoporosis.
Bone mineral density (BMD) was reported in Hartkamp 2004. This was assessed at the lumbar spine in all participants at baseline and six months and twelve months. At baseline BMD was 0.987 gm/cm2 in the DHEA group and 1.009 gm/cm2 in the placebo group. After 12 months there was no significant difference in BMD from baseline of in terms of change between the two treatment groups (DHEA at 12 months 0.992 versus placebo 1.009). Menopausal status and presence of other medications aimed at improving BMD appeared to impact on the effect of DHEA, but numbers in each group were small. No data was presented about the impacted base on steroid experience, but the authors state that there was no significant difference in BMD depending on whether currently on steroids or not. It should be noted that the primary outcome in this study was reported to be well‐being and fatigue. No data was presented for these outcomes.
Van Vollenhoven 1999 reported BMD in all 21 participants at six months and reported a significant reduction in BMD at the lumbar spine in those on placebo versus no significant change in the DHEA group (data presented in graphical form only).
From the 381 participants in Petri 2004, a subgroup of 55 participants continuously receiving steroids for at least six months had BMD measured at baseline and again at six months (a further 11 underwent baseline examination but did not attend for follow up because of withdrawal from the study ‐ they were not included in the analysis). The authors report that BMD was statistically significantly improved in the DHEA treatment group as compared to placebo at both the lumbar spine (DHEA: +1.7% versus placebo: ‐1.1%; P = 0.003) and hip (DHEA: 2% versus placebo ‐0.3%; P = 0.013)
Nordmark 2005 reported limited BMD results in their trial of low dose DHEA. They found no increase in BMD at either lumbar spine, hip or total body measured by dual energy x‐ray absorptiometry (DEXA) scanning in the group receiving DHEA but no other details were given.
Additional outcome measure For completeness, we report the findings of three trials that used different composite clinical measures.
Assessment of disease activity in SLE is difficult both in clinical practice and in studies. A number of the studies (Van Vollenhoven 1999; Petri 2002; Petri 2004) have reported on the use of composite scores or responder indices based on a range of different measures in each trial.
Petri 2002 based their definition of responder on maintaining a dose of prednisolone of 7.5 mg per day or less for at least two months. All participants on the trial had to be on a dose of 10 to 30 mg of prednisolone at the outset of the trial. They did not report a statistically significant higher proportion of responders among those treated with DHEA, when compared to placebo (% responders: placebo ‐ 41%; 100 mg ‐ 44%; 200 mg ‐ 55% P = 0.1). Prior to unblinding the trial the baseline characteristics of the participants were reviewed and the responder rate recalculated on the basis of level of disease activity as assessed by the SLEDAI, with participants being stratified into mild disease (SLEDAI = 2) and more active disease (SLEDAI > 2). The more active group accounted for 72% of all participants. The reasoning behind this redesign of the protocol was the high rate of 'responders' in the group with low baseline activity on the SLEDAI regardless of whether on placebo or DHEA 100 mg or DHEA 200 mg. On re‐analysis of only those with a SLEDAI greater than 2, 29% of those on placebo "responded" versus 38% of those on 100 mg DHEA and 51% on 200 mg DHEA per day (200 mg versus placebo was statistically significant; P = 0.03).
One study (Petri 2004) a priori defined responders as participants who experienced no clinical deterioration and had improvement or stabilisation over the duration of the study in two disease activity measures (SLEDAI and SLAM) and two quality of life measures (patient's global assessment and KFSS). A total of 381 participants were recruited initially, but while the trial was progressing, data from an earlier trial resulted in a modification of the inclusion criteria (Petri 2002). While the trial was still blinded, a subgroup was identified as those with a SLEDAI > 2. Overall, no significant difference was reported in responder rates with 42% response among those receiving placebo versus 51% among those receiving DHEA. In the sub‐group with SLEDAI > 2 a statistically significant difference in percent of responders was reported; 45% responded in the placebo group and 59% in the DHEA group (P = 0.02).
Van Vollenhoven 1999 defined responders by stabilisation of the major lupus manifestation at the six‐month time point giving specific definitions for stabilisation for renal, haematologic and serositis manifestations. Seven of the nine participants receiving DHEA were classified as responders as compared to four of ten who received placebo.
Subgroup analysis We identified six subgroups that would be of clinical importance in terms of exploring effect size differences for primary outcomes:
1. Type of DHEA analogue: Only one DHEA analogue was used in the studies reported to date so no subgroup analysis was appropriate
2. Dose: Only one study compared different doses of DHEA (Petri 2002). Sixty three participants received 100 mg DHEA and 64 received 200 mg DHEA. This study did not report any of the primary outcome measures we identified other than adverse events (Table 5). There were slightly more withdrawals due to adverse events as the dose increased (placebo: 5%; 100 mg: 6%; 200 mg: 9%). Androgen related adverse events (hirsutism, acne, menorrhagia) were not higher among those treated with 200 mg when compared to those receiving 100 mg doses. One other study used a 100 mg dose of DHEA (Van Vollenhoven 1995). This was a small study of 30 participants but it did demonstrate improvements in both SLEDAI and patient global scores. Androgenic adverse events were reported with acne being described in 57% of those receiving DHEA.
One study used the lower dose of DHEA of 10 to 30mg twice a day. It reported positive quality of life benefits from DHEA for some of the SF36 domains but no effect on disease activity.
3. Baseline disease severity: None of the trials allowed within study comparison of severity of disease. There is no universally accepted classification of severity and definition varied between trials. One study only included participants with severe disease and the results for this group have been presented separately Van Vollenhoven 1999.
4. Age: results were not reported by age bands therefore it was not possible to undertake any subgroup analysis.
5. Gender: Only one of the studies did not exclude males, however, only 3 of the 19 participants recruited were male. Results were not reported separately for the males, and all three received placebo.
6. Race: The predominant race reported by four of the trials (Nordmark 2005 did not report race) was Caucasian and none of these studies reported results separately for different ethnic groups (Van Vollenhoven 1995; Petri 2002; Hartkamp 2004; Petri 2004). The Van Vollenhoven 1999 was not predominantly Caucasian but little detail was given about race and the results were not reported separately. One study (Chang 2002) was conducted exclusively in Chinese women. This study did not report a positive benefit from DHEA in terms of disease activity (as measured by SLEDAI or SLAM) or quality of life. There may be some differences in the reporting or occurrence of adverse events that are related to race/culture. Very few androgenic adverse events were reported in this study, other than acne.
Sensitivity analysis A priori we indicated four sensitivity analyses that would be relevant. There was very little overlap in the studies to enable meta‐analysis, either in terms of the disease severity or outcomes reported. For no outcome was it possible to include more than two trials in any given disease severity subgroup in a meta analysis. No sensitivity analysis was undertaken.
A summary of the meta‐analyses of benefits and harms are presented in clinical relevance tables (Table 6;Table 7).
6. Clinical Relevance Table ‐ Summary of Meta‐analysis: Benefits.
| Outcome | # patients(# trials) | Control baseline m | Wt absolute change | Relative % change | NNT (B) or NNT (H) | Statistical Sig | Quality of Evidence |
| SLEDAI (mild/moderate) (0‐105) | 148(2) | 6.55* | 0.6% reduction (0.6 points less on a scale of 0‐105) | 9.3% improvement | NA | not statistically significant | Gold |
| 95% confidence interval | ‐2.12 to 0.89 | ||||||
| SLEDAI (severe) (0‐105) | 19(1) | 9.4 | 6% reduction (6.4 points less on a scale of 0‐105) | 68% improvement | 3 | borderline statistically significant | Silver |
| 95% confidence interval | ‐13.12 to 0.32 | ||||||
| HRQoL (mild/moderate) Patient Global (0‐100) | 148 (2) | 28.5 | 11.5% reduction (11.5 point reduction on a scale of 0‐100) | 40.4% improvement | 5 | statistically significant | Gold |
| 95% confidence interval | ‐19.1 to ‐3.8 | ||||||
| HRQoL (severe) (0‐100) | 19 (1) | 52.6 | 0.2% reduction (0.2 point reduction on a scale of 0‐100) | 0.4% improvement | NA | not statistically significant | Silver |
| 95% confidence interval | ‐3.7 to 3.3 | ||||||
| Legend: | SLEDAI ‐ SLE disease activity index HRQOL ‐ Health Related Quality of Life | NA=not applicable |
7. Clinical Relevance Table: Summary Meta‐analysis table: adverse events.
| Outcome | # patients (#trials) | Event rate (placebo) | Event rate (treated) | Relative risk | Absolute risk dif | NNH |
| Adverse Events (mild/moderate): Acne | 657 (4) | 56/326 (17.2%) | 123/331 (37.2%) | 2.2 | 0.2 | 5 |
| 95% confidence interval | 1.65 to 2.83 | 0.13 to 0.26 | ||||
| Adverse Events (mild/moderate): Menstrual Change | 276 (3) | 5/137 (3.6%) | 6/139 (4.3%) | 1.2 | 0.1 | not statistically significant |
| 95% confidence interval | 0.38 to 3.77 | ‐0.04 to 0.05 | ||||
Discussion
SLE is a complex multiorgan autoimmune condition characterised by flares and remissions. Its impact on individuals' physical and psychological well being is variable. In the absence of a curative treatment, existing therapies aim to control flares and limit end organ damage. As such an individual can be on treatments for many years. At the cornerstone of current therapy are corticosteroids and with them the well recognised adverse event profile. The aim of new therapies, therefore, may not only be to control symptoms and disease progression but also to reduce the dependence on steroid treatment.
This review identified a total of seven RCTs evaluating the effectiveness of DHEA in the treatment of SLE. In keeping with the complex natural history of SLE, the outcome measures assessed in the trials were diverse and reported variably. There is no single accepted measure of SLE disease activity. It has, therefore, been difficult to draw conclusions about the effectiveness of DHEA in the treatment of SLE.
We found evidence of DHEA use in three dose regimens: 200 mg once daily; 100 mg once daily and in a low dose regimen of 10 to 30mg per day.
OMERACT defined three domains that need to be considered when evaluating effectiveness of treatments for SLE: Disease activity; Health related quality of life and adverse events.
For mild to moderate disease, four of the five studies reporting SLEDAI as a measure of disease activity, including the two trials that could be combined in meta analysis, supported the conclusion that there was 'gold' ranking evidence of no difference in change in SLEDAI score when participants were treated with DHEA as compared to placebo. However, one well conducted study did find a greater proportion of patents on DHEA experienced stabilisation or improvement in disease activity as measured by SLEDAI (8.3% more participants on DHEA; P = 0.04) . The one very small study in participants with severe SLE found a borderline statistically significant improvement in SLEDAI. Those studies reporting disease activity measured by SLAM provided 'gold' ranking evidence of no benefit from DHEA when compared to placebo. Similarly, there is currently no evidence of a reduction in end organ damage.
HRQoL measured by various questionnaires including SF36 was not found to be improved significantly by DHEA, except in two domains of the SF36 questionnaire tool. Most studies used Patient Global Assessment to measure quality of life. A statistically significant reduction in Patient Global Assessment was observed consistently in the four studies reporting this outcome. The minimum clinically significant change is generally considered to be 10% (10mm on the 0 to 100mm scale). The meta‐analysis estimate of a mean reduction of 11.5% was therefore of clinical significance. Physician Global Assessment was reported in three studies and no statistically significant benefit was reported when compared to placebo.
Predictable androgenic adverse events were the most commonly reported side effects of DHEA. No conclusions could be drawn about longer term potential adverse effects, for example cardiovascular complications or malignancy or rare adverse events, because of the relatively small number of trial participants with SLE to date and the short follow up experience reported in all of the trials. It is our intention to include data from observational studies and case series about adverse events at the next update of this review.
Other outcomes Much attention has been given to the potential steroid sparing role of drugs such as DHEA in SLE. Two of the three studies reporting this outcome did not find a statistically significant difference in BMD from placebo. The third study, on a small subgroup of a larger study, did report small percentage improvements in BMD in the DHEA treated group compared with placebo.
Composite scores More than sixty different measures of disease activity have been described in SLE studies since the 1980s. The measures in routine clinical use were largely designed in the context of longitudinal observational studies rather than as outcome measures for randomised controlled trials (Strand 1999). Reflecting the complex nature of the condition and its fluctuating natural history, some authors have attempted to describe disease activity response in terms of composite measures. Improvements across these combined measures are then used to define "responders" within the clinical studies. Whilst there may be good reasons for these composite scores and responder indices, unlike the SLEDAI, SLAM, ECLAM, LAI or BILAG, they are, as yet, unvalidated. Although these composite indices have some appeal, their utility is unclear in the absence of their prospective validation in randomised controlled trials (Strand 2004) Guidance from the FDA suggests that any responder index should be assessed for reliability, face validity, content validity and sensitivity to change. At present, there are no generally accepted and validated responder indices in lupus (CDER 2005). Earlier attempts to provide composite measures of disease activity, such as the physicians' visual analogue scale, have been shown to be disappointingly unreliable in clinical practice (Wollaston 2004). At present the use of composite disease responder indices must be viewed with some caution.
Limitations of review The major limitation for this review was the lack of consensus around measures of disease activity. As such it has been difficult to draw conclusions from the trials to date or to meta‐analyse results. Trials have also tended to involve small numbers of largely Caucasian females. This limits the generalisability of the results to other groups. Short term follow up means that uncertainty remains around safety of these androgenic drugs. Furthermore, a number of the trials excluded participants from the analysis who experienced early adverse events or withdrew in the early stages of the study. This substantially weakens the study design and reduces confidence in the conclusions reported by the authors. Finally, changes in the definition of "active disease" during the course of two of the trials (Petri 2002; Petri 2004) and subsequent subgroup analysis complicates the interpretation of these trials.
From a methodological perspective, we have systematically searched a wide range of published literature resources. We have not, however, directly contacted the drug manufacturers or trialists to access additional information.
Authors' conclusions
Implications for practice.
From the seven RCTs to date there is 'gold' ranking evidence that DHEA: * had little clinical effect on disease activity in those with mild/moderate disease (measured by SLEDAI or SLAM) but one study demonstrated evidence of stabilisation or improvement in 8.3% more participants than those treated with placebo. * had a modest but clinically significant improvement in health related quality of life measured by Patient Global Assessment, estimated as 11.5% (11.5 mm on a 100mm scale) by meta‐analysis. * resulted in a greater number of participants experiencing adverse events, particularly androgenic effects such as acne where participants risk was doubled when compared to placebo (RR 2.2; 95% CI 1.65 to 2.83)
Long term outcomes and safety remain unstudied.
The difficulties in studying effectiveness of DHEA for SLE highlight the difficulties of studying any treatment for a disease as complex as SLE. A clinical consensus on meaningful outcomes needs to be supported by scientific evaluation of the reliability and validity of measures of disease activity.
Implications for research.
We have identified the following key areas for research for DHEA in the treatment of SLE: * assessment of long term outcomes including safety; * consensus on the clinical definition of disease activity and severity; * evaluation of the reliability and validity of composite measures in SLE.
What's new
| Date | Event | Description |
|---|---|---|
| 16 September 2008 | Amended | Converted to new review format. CMSG ID C027‐R |
Acknowledgements
We would like to thank Prof Norman Waugh for his help in identifying the topic for the review and contribution to the methods.
Data and analyses
Comparison 1. Disease Activity.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 SLEDAI | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Mild to moderate SLE | 2 | 148 | Mean Difference (IV, Fixed, 95% CI) | ‐0.61 [‐2.12, 0.89] |
| 1.2 Severe SLE | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐6.4 [‐13.12, 0.32] |
1.1. Analysis.
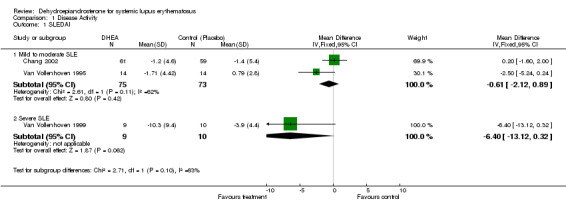
Comparison 1 Disease Activity, Outcome 1 SLEDAI.
Comparison 2. Quality of Life.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Patient Global | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Mild to Moderate SLE | 2 | 148 | Mean Difference (IV, Fixed, 95% CI) | ‐11.46 [‐19.08, ‐3.84] |
| 1.2 Severe SLE | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐3.67, 3.27] |
| 2 Physician Global | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Mild to moderate SLE | 2 | 148 | Mean Difference (IV, Fixed, 95% CI) | ‐3.16 [‐8.12, 1.80] |
| 2.2 Severe SLE | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐9.80 [‐40.36, 20.76] |
2.1. Analysis.
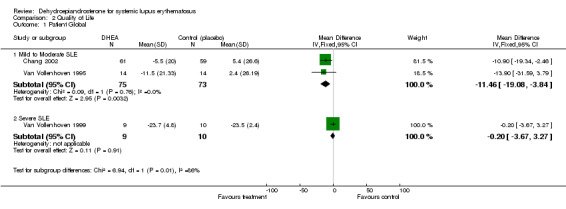
Comparison 2 Quality of Life, Outcome 1 Patient Global.
2.2. Analysis.

Comparison 2 Quality of Life, Outcome 2 Physician Global.
Comparison 3. Adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Acne | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Mild to moderate SLE | 4 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.16 [1.65, 2.83] |
| 1.2 Severe SLE | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.68, 5.85] |
| 2 Hirsutism | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Mild to moderate SLE | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Severe SLE | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Menstrual change | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Mild to moderate SLE | 3 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.38, 3.77] |
| 3.2 Severe SLE | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.98, 7.22] |
3.1. Analysis.
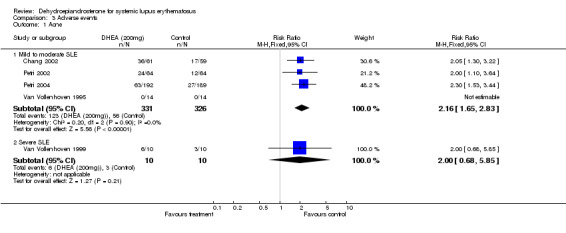
Comparison 3 Adverse events, Outcome 1 Acne.
3.2. Analysis.
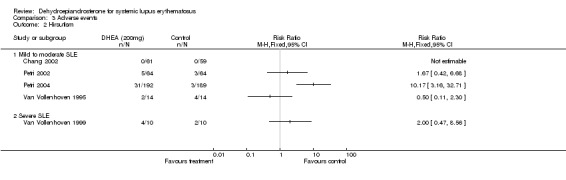
Comparison 3 Adverse events, Outcome 2 Hirsutism.
3.3. Analysis.
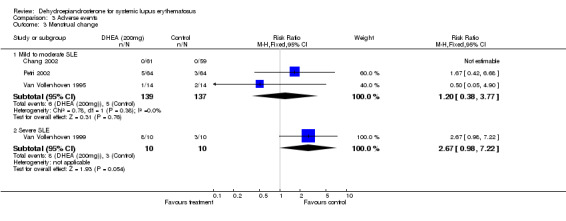
Comparison 3 Adverse events, Outcome 3 Menstrual change.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Chang 2002.
| Methods | double blind RCT 6 months | |
| Participants | Total: 120 Sex: female Ethnicity: Chinese Median age: not stated Inclusion criteria:active SLE (including SLEDAI > 2), on <10mg prednisolone Exclusion criteria:other immunosuppressants | |
| Interventions | Grp 1: DHEA 200mg/dy Grp 2: placebo | |
| Outcomes | Stated primary:
SLAM SLEDAI, HRQoL, flares |
|
| Notes | Funding: Genelabs + National Science Council | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Hartkamp 2004.
| Methods | Double blind, controlled trial (Probably randomised but does not specify) 12 months | |
| Participants | Total: 60 Sex: female Ethnicity: 82‐93% Caucasian Median age: 41‐45 years Inclusion criteria: quiescent SLE, on <10mg prednisolone Exclusion criteria: nil else | |
| Interventions | Grp 1: DHEA 200mg/dy Grp 2: placebo | |
| Outcomes | Stated primary: Wellbeing and fatigue (not reported) SLEDAI, Bone mineral density |
|
| Notes | Funding: Dutch Arthritis Association | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Nordmark 2005.
| Methods | Double blind RCT 6 months (open label continuation for further 6 months) | |
| Participants | Total: 37* (4 dropped out and not included in analysis, 41 recruited) Sex: female Ethnicity: not stated Mean age: 47 to 48 years (selection involved stratification into 2 age group of equal size) | |
| Interventions | Grp 1: DHEA 20‐30mg/dy Grp 2: placebo | |
| Outcomes | Stated primary: HRQoL and behaviour (various tools) SLEDAI; SLICC; Bone mineral density; biological markers; |
|
| Notes | Funding: various non industry sources | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Petri 2002.
| Methods | double blind RCT 7‐9 months | |
| Participants | Total: 191 Sex: female Ethnicity: 55‐57% Caucasian; 25‐27% African‐American Median age: 30‐41yr Inclusion criteria: >10mg prednisolone for 12mths Exclusion criteria: other immunosuppressants | |
| Interventions | Grp 1: DHEA 100mg/dy Grp 2: DHEA 200mg/dy Grp 3: placebo | |
| Outcomes | Stated primary: 'Responder'
days of prednisolone <7.5mg SLEDAI (not reported), HRQoL (not reported), Fatigue, (not reported), SLICC (not reported) biological markers |
|
| Notes | Funding: Genelabs Technologies Inc | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Petri 2004.
| Methods | double blind RCT 12 months | |
| Participants | Total: 381 Sex: female Ethnicity:71‐77% Caucasian Median age:44 years Inclusion criteria: active SLE Exclusion criteria: | |
| Interventions | Grp 1: DHEA 200mg/dy Grp 2: placebo | |
| Outcomes | Stated primary: "responders" on composite score SLEDAI, HRQoL, flares, SLAM, fatigue (BMD in subgroup) |
|
| Notes | Funding: Genelabs Technologies Inc | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Van Vollenhoven 1995.
| Methods | double blind RCT 3 months | |
| Participants | Total: 28* (2 dropped out before study started and not included in analysis ‐ 30 recruited) Sex: female Ethnicity: 64‐79% Caucasian; 21‐36% other Median age: 35‐40 years Inclusion criteria: mild to moderate SLE Exclusion criteria: renal disease, high dose steroids or other immunosuppressants | |
| Interventions | Grp 1: DHEA 100mg/dy Grp 2: placebo | |
| Outcomes | Stated primary:
not stated SLEDAI, prednisolone dose, HRQoL, Physician global VAS, flares, other biochemical markers |
|
| Notes | Funding: Northern California Arthritis Foundation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Van Vollenhoven 1999.
| Methods | double blind RCT 6 months | |
| Participants | Total: 19 Sex: male or female (only 3 male) Ethnicity: 33‐40% Caucasian; 11‐20% African‐American; 40‐44% other Median age:35‐39 yr Inclusion criteria: severe SLE Exclusion criteria: | |
| Interventions | Grp 1: DHEA 200mg/dy Grp 2: placebo | |
| Outcomes | Stated primary: stabilise major lupus manifestation SLEDAI, SLAM, HRQoL, renal proteinuria, biological SLEDAI, Prednisolone dose, Patient global VAS, Physician global VAS, Bone mineral density, other biochemical markers |
|
| Notes | Funding: NIH grant | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
HRQoL ‐ health related quality of life including SF36 and Patient Visual analogue scores
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Barry 1998 | Case series; no control group |
| Chang 2004 | Outcome measure not included in this review |
| van Vollenhoven 1992 | Case series; no control group |
| van Vollenhoven 1993 | Case series; no control group |
| van Vollenhoven 1994 | Case series; no control group |
| van Vollenhoven 1996 | Case series; no control group |
| van Vollenhoven 2001 | Study less than 3 months duration; placebo controlled trial |
| vanVollenhoven 1994b | Case series; no control group |
Contributions of authors
CB and DC had the original idea for the work following work initiated by the HTA program. CB drafted the protocol. DC, PR, LM and ST undertook the reviewing, commented on drafts and approved the final version.
Sources of support
Internal sources
Department of Public Health, University of Aberdeen, UK.
External sources
Health Technology Assessment program (via TAR budget), UK.
Declarations of interest
There are no known conflicts of interest.
Edited (no change to conclusions)
References
References to studies included in this review
Chang 2002 {published data only}
- Chang DM, Lan JL, Lin HY, Luo SF. Dehydroepiandrosterone treatment of women with mild‐to‐moderate systemic lupus erythematosus: a multicenter randomized, double‐blind, placebo‐controlled trial. Arthritis and Rheumatism 2002;46(11):2924‐7. [DOI] [PubMed] [Google Scholar]
Hartkamp 2004 {published data only}
- Hartkamp A, Geenen R, Godaert GL, Bijl M, Bijlsma JW, Derksen RH. The effect of dehydroepiandrosterone on lumbar spine bone mineral density in patients with quiescent systemic lupus erythematosus. Arthritis and Rheumatism 2004;50(11):3591‐5. [DOI] [PubMed] [Google Scholar]
Nordmark 2005 {published data only}
- Nordmark G, Bengtsson C, Larsson A, Karlsson FA, Sturfelt G, Ronnblom L. Effects of dehydroepiandrosterone supplement on health‐related quality of life in glucocorticoid treated female patients with systemic lupus erythematosus. Autoimmunity 2005;38(7):531‐40. [DOI] [PubMed] [Google Scholar]
Petri 2002 {published data only}
- Petri MA, Lahita RG, Vollenhoven RF, Merrill JT, Schiff M, Ginzler EM, Strand V, Kunz A, Gorelick KJ, Schwartz KE, ‐Study‐Group. Effects of prasterone on corticosteroid requirements of women with systemic lupus erythematosus: a double‐blind, randomized, placebo‐controlled trial. Arthritis and Rheumatism 2002;46(7):1820‐9. [DOI] [PubMed] [Google Scholar]
Petri 2004 {published data only}
- Mease PJ, Ginzler EM, Gluck OS, Schiff M, Goldman A, Greenwald M, Cohen S, Egan R, Quarles BJ, Schwartz KE. Effects of prasterone on bone mineral density in women with systemic lupus erythematosus receiving chronic glucocorticoid therapy. Journal of Rheumatology 2005;32(4):616‐21. [PubMed] [Google Scholar]
- Petri MA, Mease PJ, Merrill JT, Lahita RG, Iannini MJ, Yocum DE, Ginzler EM, Katz RS, Gluck OS, Genovese MC, Vollenhoven R, Kalunian KC, Manzi S, Greenwald MW, Buyon JP, Olsen NJ, Schiff MH, Kavanaugh AF, Caldwell JR, Ramsey‐Goldman R, Clair EW, Goldman AL, Egan RM, Polisson RP, Moder KG, Rothfield NF, Spencer RT, Hobbs K, Fessler BJ, Calabrese LH, Moreland LW, Cohen SB, Quarles BJ, Strand V, Gurwith M, Schwartz KE. Effects of prasterone on disease activity and symptoms in women with active systemic lupus erythematosus. Arthritis and Rheumatism 2004;50(9):2858‐68. [DOI] [PubMed] [Google Scholar]
Van Vollenhoven 1995 {published data only}
- Vollenhoven RF, Engleman EG, McGuire JL. Dehydroepiandrosterone in systemic lupus erythematosus. Results of a double‐blind, placebo‐controlled, randomized clinical trial. Arthritis and Rheumatism 1995;38(12):1826‐1831. [DOI] [PubMed] [Google Scholar]
- vanVollenhoven RF, Engleman EG, Lambert RE, Lee YSL, McGuire JL. Treatment of Systemic Lupus‐Erythematosus with Dehydroepiandrosterone ‐ Interim Analysis of A Double‐Blinded, Randomized, Placebo‐Controlled, Clinical‐Trial. Arthritis and Rheumatism 1993;36(9 Suppl S Sep):S92. [Google Scholar]
- vanVollenhoven RF, Lambert RE, Lee YSL, McGuire JL, Engleman EG. Treatment of Systemic Lupus‐Erythematosus with Dehydroepiandrosterone Follow‐Up from An Open‐Label Clinical‐ Trial. Arthritis and Rheumatism 1993;36(9 Suppl S):S228. [Google Scholar]
Van Vollenhoven 1999 {published data only}
- Vollenhoven RF, Park JL, Genovese MC, West JP, McGuire JL. A double‐blind, placebo‐controlled, clinical trial of dehydroepiandrosterone in severe systemic lupus erythematosus. Lupus 1999;8(3):181‐187. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Barry 1998 {published data only}
- Barry NN, McGuire JL, Vollenhoven RF. Dehydroepiandrosterone in systemic lupus erythematosus: Relationship between dosage, serum levels, and clinical response. Journal of Rheumatology 1998;25(12):2352‐6. [PubMed] [Google Scholar]
Chang 2004 {published data only}
- Chang DM, Chu SJ, Chen HC, Kuo SY, Lai JH. Dehydroepiandrosterone suppresses interleukin 10 synthesis in women with systemic lupus erythematosus. Annals of Rheumatic Diseases 2004;63(12):1623‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Vollenhoven 1992 {published data only}
- Vollenhoven RF, Lee YSL, Lambert RE, Engleman EG, Mcdevitt HO, McGuire JL. Treatment of Systemic Lupus‐Erythematosus with Dehydroepiandrosterone ‐ A Pilot‐Study. Arthritis and Rheumatism 1992;35(9 Suppl S):S55. [Google Scholar]
van Vollenhoven 1993 {published data only}
- Vollenhoven RF, Lambert RE, Lee YSL, McGuire JL, Engleman EG. Treatment of Systemic Lupus‐Erythematosus with Dehydroepiandrosterone Follow‐Up from An Open‐Label Clinical‐ Trial. Arthritis and Rheumatism 1993;36(9 Suppl S):S228. [Google Scholar]
van Vollenhoven 1994 {published data only}
- Vollenhoven RF, Engleman EG, McGuire JL. An open study of dehydroepiandrosterone in systemic lupus erythematosus. Arthritis and Rheumatism 1994;37(9):1305‐10. [DOI] [PubMed] [Google Scholar]
van Vollenhoven 1996 {published data only}
- Vollenhoven RF. Increased bone mineral density in patients with SLE treated with DHEA. Arthritis and Rheumatism 1996;39(9 Suppl S Sep):1588. [Google Scholar]
van Vollenhoven 2001 {published data only}
- Vollenhoven RF. Dehydroepiandrosterone improves cognitive function in patients with systemic lupus erythematosus: Results of a double‐blinded, placebo controlled pilot study. Lupus 2001;10(Supplement 1):S51. [Google Scholar]
vanVollenhoven 1994b {published data only}
- Vollenhoven R, Morabito L, Engleman E, Lambert R, Lee Y, S, McGuire J, L. Treatment of systemic lupus erythematosus with dehydroepiandrosterone, two‐year follow‐up from an open‐label clinical trial. Arthritis and Rheumatism 1994;37(9 SUPPL.):S407. [Google Scholar]
Additional references
Boumpas 1992
- Boumpas DT, Austin HA, Vaughn E, Klippel JH, Steinberg AD, Yarboro CH, Barlow JE. Controlled Trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet 1992;340:741‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Caccavo 1997
- Caccavo D, Lagana B, Mitterhofer AP, Ferri GM, Afeltra A, Amoroso A, Bonomo L. Long‐term treatment of systemic lupus erythematosus with cyclosporin A. Arthritis and Rheumatism 1997;40:27‐35. [DOI] [PubMed] [Google Scholar]
Cates 2003
- Dr Chris Cates. EBM Website. www.nntonline.net 2003.
CDER 2005
- Center for Drug Evaluation and Research (CDER). Guidance for Industry: Systemic Lupus Erythematosus ‐ Developing Drugs for Treatment. U.S. Department of Health and Human Services UFood and Drug Administration, U.S. Department of Health and Human Services. U.S. Department of Health and Human Services, 2005.
CHSG 1991
- Canadian Hydroxychloroquine Study Group. A randomized study of the effect of withdrawing hydroxychloroquine sulphate in systemic lupus erythematosus. New England Journal of Medicine 1991;324:150‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Derksen 1998
- Derksen RHWM. Dehydroepiandrosterone (DHEA) and systemic lupus erythematosus. Seminars in Arthritis and Rheumatism 1998;27:335‐47. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
FitzGerald 1999
- FitzGerald JD, Grossman JM. Validity and reliability of retrospective assessment of disease activity and flare in observational cohorts of lupus patients. Lupus 1999;8(8):638‐44. [DOI] [PubMed] [Google Scholar]
Genelabs 2001
- Genelabs Technologies I. GL701 (DHEA, prasterone) for the treatment of systemic lupus erythematosus (SLE) in women. Briefing Document FDA Arthritis Advisory Committee. Briefing Document, FDA Arthritis Advisory Committee 2001; Vol. Briefing Document, FDA Arthritis Advisory Committee. [MEDLINE: ]
Hart 1983
- Hart HH, Grigor RR, Caughey DE. Ethnic difference in the prevalence of systemic lupus erythematosus. Annals of Rheumatic Diseases 1983;42(5):529‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2006
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions 4.2.6 [updated September 2006]. In: The Cochrane Library, Issue 4, 2006. John Wiley & Sons, Ltd. Chichester, UK.
Homik 2004
- Homik J, Cranney A, Shea B, Tugwell P, Wells G, Adachi R, Suarez‐Almazor M. Bisphosphonates for steroid induced osteoporosis. Cochrane Database of Systematic Reviews 1999, Issue 1. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, McQuay HJ. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI] [PubMed] [Google Scholar]
Johnson 1995
- Johnson AE, Gordon C, Palmer RG, Baethge BA. The prevalence and incidence of systemic lupus erythematosus in Birmingham, England. Relationship to ethnicity and country of birth. Arthritis and Rheumatism 1995;38(4):551‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Johnson 1996
- Johnson AE, Gordon C, Hobbs FDR, Baethge BA. Undiagnsoed systemic lupus erythematosus in the community. Lancet 1996;347:367‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Khan 2000
- Khan KS, ter Riet G, Glanville J, Sowden A, Kleijnen J. Undertaking systematic reviews of research on effectiveness: CRD's guidance for those carrying out and commissioning reviews. CRD Report 4 (2nd edition) 2000; Vol. CRD Report 4 (2nd edition).
Kingdon 2001
- Kingdon EJ, McLean AG, Psimenou E, Davenport A, Powis SH, Sweny P, Burns A. The safety and efficacy of MMF in lupus nephritis: a pilot study. Lupus 2001;10(9):606‐11. [DOI] [PubMed] [Google Scholar]
Lee 1993
- Lee SS, Li CS, Li PCK. Clinical profile of Chinese patients with systemic lupus erythematosis. Lupus 1993;2:105‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
McCarty 1995
- McCarty DJ, Manzi S, Medsger TA Jr, Ramsey‐Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis and Rheumatism 1995;38(9):1260‐70. [DOI] [PubMed] [Google Scholar]
Mok 2003
- Mok CC, Lau CS. Lupus in Hong Kong Chinese. Lupus 2003;12(9):717‐22. [DOI] [PubMed] [Google Scholar]
Nived 1997
- Nived O, Sturfelt G. In: Isenberg D.A, Tucker L.B editor(s). Does the Black population in Africa get SLE? If not why not. London: Martin Dunitz Ltd, 1997. [MEDLINE: ] [Google Scholar]
Petri 2005
- Petri M, Kim MY, Kalunian KC, Grossman J, Hahn BH, Sammaritano LR, Lockshin M, Merrill JT, Belmont HM, Askanase AD, McCune WJ, Hearth‐Holmes M, Dooley MA, Feldt J, Friedman A, Tan M, Davis J, Cronin M, Diamond B, Mackay M, Sigler L, Fillius M, Rupel A, Licciardi F, Buyon JP. Combined oral contraceptives in women with systemic lupus erythematosus. New England Journal of Medicine 2005;353(24):2550‐8. [DOI] [PubMed] [Google Scholar]
Ruiz‐Irastorza 2001
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Silman 1997
- Silman A, Shipley M. Opthalmological monitoring for hydroxychloroquire toxicity: a scientific review of the available data. British Journal of Rheumatology 1997;36:599‐601. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Singh 1999
- Singh G, Triadafilopoulos G. Epidemiology of NSAUD‐induced gastrointestinal complications. Journal of Rheumatology 1999;26(Suppl 56):18‐24. [MEDLINE: ] [PubMed] [Google Scholar]
Strand 1999
- Strand V, Gladman D, Isenberg D, Petri M, Smolen J, Tugwell P. Outcome measures to be used in clinical trials in systemic lupus erythematosus. Journal of Rheumatology 1999;26(2):490‐7. [PubMed] [Google Scholar]
Strand 1999b
- Strand V. Biologic agents and innovative interventional approaches in the management of systemic lupus erythematosus. Current Opinion in Rheumatology 1999;11(5):330‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Strand 2004
- Strand V. Clinical trial design in systemic lupus erythematosus: lessons learned and future directions. Lupus 2004;13(5):406‐11. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tugwell 2004
- Tugwell P, Shea B, Boers M, Brooks P, Simon L, Strand V, Wells G (Editors). Evidence‐based Rheumatology. London: BMJ Books, 2004. [Google Scholar]
Whelton 2000
- Whelton A. Renal and related cardiovascular effects of conventional and COX‐2‐specific NSAIDs and non‐NSAID analgesia. American Journal of Therapeutics 2000;7:63‐74. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wollaston 2004
- Wollaston SJ, Farewell VT, Isenberg DA, Gordon C, Merrill JT, Petri MA, Kalunian KC. Defining response in systemic lupus erythematosus: a study by the Systemic Lupus International Collaborating Clinics group. Journal of Rheumatology 2004;31(12):2390‐4. [PubMed] [Google Scholar]