

Abstract
The adverse consequences of developmental exposures to perfluorooctanoic acid (PFOA) are established in mice, and include impaired development of the mammary gland (MG). However, the relationships between timing or route of exposure, and consequences in the MG have not been characterized. To address the effects of these variables on the onset and persistence of MG effects in female offspring, timed pregnant CD-1 dams received PFOA by oral gavage over various gestational durations. Cross-fostering studies identified the 5 mg/kg dose, under either lactational- or intrauterine-only exposures, to delay MG development as early as postnatal day (PND) 1, persisting beyond PND 63. Intrauterine exposure during the final days of pregnancy caused adverse MG developmental effects similar to that of extended gestational exposures. These studies confirm a window of MG sensitivity in late fetal and early neonatal life, and demonstrate developmental PFOA exposure results in early and persistent MG effects, suggesting permanent consequences.
Keywords: Perfluorooctanoic acid (PFOA), Mammary gland, Prenatal exposure, Neonatal exposure, Lactation, Dosimetry, Delayed development, Fetal origins of adult disease
1. Introduction
Perfluorooctanoic acid (PFOA) is a broadly used industrial compound, as well as a final environmental degradation product of many other perfluorinated compounds. Numerous applications of PFOA arise from its capacity to resist extreme temperatures and stresses, and include industrial production of fire-fighting and flame retardant materials, water- and oil-repellant coatings for fabrics and food packaging, surfactants, paint additives, and electrical insulation, among many others. However, the chemical properties of PFOA, which lend so well to these commercial applications, result also in its environmentally persistent nature [1]. Given its commercial and environmental ubiquity, in conjunction with its persistence, it is not surprising that PFOA has been detected in the sera of humans and wildlife, and found to be widely distributed by a number of survey studies [2–9]. Recent estimates suggest that the non-occupationally exposed American exhibits an average serum PFOA concentration of 3.9 ng/ml, which is down from the national average 2 years prior (5.2 ng/ml) [10]. While sources of exposure are poorly characterized, this may result from ingestion of contaminated food or water, as compared to the presumed inhalational route among the occupationally exposed. This average serum PFOA concentration, to date, has not been associated with adverse health effects in humans. However, occupationally relevant levels, higher than those observed in the average American, have been observed in animal toxicity studies [11,12]. Toxicologic studies of carcinogenesis in animals have indicated the potential for high-dose (generally, >10 mg/kg/day in rodents, chronically), adult PFOA exposures to result in hepatotoxicity marked by extreme hypertrophy, as well as a common tumor triad consisting of hepatocellular carcinomas, pancreatic acinar-cell tumors, and Leydig cell tumors [13,14].
Recent developmental toxicity studies in an outbred mouse strain have identified the capacity for PFOA to hinder early life body weight gain in gestationally exposed offspring [11,15,16], as well as delay mammary gland (MG) development in female offspring [17] independent of body growth retardation. Treatment of pregnant dams with 5 mg/kg PFOA on gestation days (GD) 12–17 was demonstrated as sufficient to produce developmental delays in the 10- and 20-day-old offspring MG [17]. Whether this response is dependent on use of this relatively high exposure compared to that received by humans, is specific to late-pregnancy timing, or requires both in utero and lactational exposures, is not known. Nevertheless, these routes of exposure have immediate relevance to human health, as PFOA has been detected in both the cord blood and breast milk of humans [18–24]. Given these potential human exposures, understanding how these changes in the MG are mediated by PFOA exposure conditions in the mouse will be critical in interpreting mode of action and health risk in human populations.
In the three studies described herein, the persistence of the MG effects present in PFOA-exposed offspring is addressed, as is the timing and route of exposure sufficient to produce these effects. Utilizing concurrent animals from previously reported studies [11], adult and late-life consequences of PFOA exposure with respect to MG tissue were examined. Then, to determine the timing of the appearance of the MG phenotype, another similar experiment was performed, addressing multiple early time points not included in the prior studies. Using the resulting data, this paper discusses the onset and duration of impaired MG development resulting from early-life PFOA exposures, the persistence of this impairment, and the subsequent late-life MG phenotype.
2. Materials and methods
2.1. PFOA
Perfluorooctanoic acid (PFOA, ammonium salt; >98% pure) was purchased from Fluka Chemical (Steinhiem, Switzerland). NMR analysis, kindly provided by 3M Company (St. Paul, MN, USA), indicated that approximately 98.9% of the chemical was straight-chain and the remaining 1.1% was branched isomers. For all studies, PFOA was dissolved by mild agitation in de-ionized water and prepared fresh daily, immediately prior to administration.
2.2. Animals
All animal studies were conducted in accordance with guidelines established by the National Health and Environmental Effects Research Laboratory Institutional Animal Care and Use Committee. Procedures and facilities were consistent with the recommendations of the 1996 NRC “Guide for the Care and Use of Laboratory Animals”, the Animal Welfare Act, and Public Health Service Policy on the Humane Care and Use of Laboratory Animals. Timed pregnant CD-1 mice were obtained from Charles River Laboratories (Raleigh, NC, USA), where females were bred overnight, and the sperm positive females, defined as GD 0, were shipped on the same day for use in these studies. Upon arrival, mice were housed individually in polypropylene cages with Alpha-dri (Shepherd Specialty Papers, Kalamazoo, MI, USA) bedding and provided pellet chow (LabDiet 5001, PMI Nutrition International LLC, Brent-wood, MO, USA) and tap water (containing PFOA at concentrations below the level of detection) ad libitum. Animal facilities were controlled for temperature (20–24 °C) and relative humidity (40–60%), and kept under a 12-h light–dark cycle.
2.3. Late-life effects cross-foster study
The study was performed in two blocks, spaced 4 weeks apart, with 56 timed pregnant mice per block (112 total). Upon arrival at the animal facility on GD 0, mice were weighed and randomly assigned to one of three treatment groups, vehicle control which received de-ionized water (n = 48), 3 mg PFOA/kg body weight (n = 28), or 5 mg PFOA/kg body weight (n = 36). On GD 1–17, mice were weighed daily and dosed by oral gavage at a 10 ml/kg volume. On GD 18–19, dams were monitored at 4-h intervals, and litters of similar ages and exposures were mixed, then fostered to yield the following seven exposure groups shown in Fig. 1A: (1) control pups nursed by control dams (control); (2) control pups nursed by dams dosed during gestation with 3 mg PFOA/kg (3L, lactational exposure); (3) control pups nursed by dams dosed during gestation with 5 mg PFOA/kg (5L, lactational exposure); (4) pups exposed in utero to 3 mg PFOA/kg nursed by control dams (3U, intrauterine exposure); (5) pups exposed in utero to 5 mg PFOA/kg nursed by control dams (5U, intrauterine exposure); (6) pups exposed in utero to 3 mg PFOA/kg nursed by dams dosed during gestation with 3 mg PFOA/kg (3U + L, intrauterine and lactational exposure); (7) pups exposed in utero to 5 mg PFOA/kg nursed by dams dosed during gestation with 5 mg PFOA/kg (5U + L, intrauterine and lactational exposure). Foster litters included 10 pups with equal representation of males and females (where possible). All pups were either assigned to foster litters or were euthanized.
Fig. 1.

Schematic of study design and implementation for (A) the late-life effects cross-foster study, (B) the early-life effects cross-foster study, and (C) the restricted-exposure study.
2.4. Early-life effects cross-foster study
The study was performed in a single block of 112 timed-pregnant CD-1 mice. Upon arrival at the animal facility on GD 0, mice were weighed and randomly assigned to one of two treatment groups, either vehicle control which received deionized water (n = 56) or 5 mg PFOA/kg body weight (n = 56), received by oral gavage. Pregnant dams were weighed daily and dosed by oral gavage at a 10 ml/kg volume on GD 8–17, as this treatment window was previously demonstrated as sufficient to impair offspring MG development without profound offspring growth deficits or postnatal loss [17]. On GD 18–19, dams were monitored at 4-h intervals, and litters of similar ages and exposures were mixed, then fostered to yield the following four exposure groups shown in Fig. 1B: (1) control pups nursed by control dams (control); (2) control pups nursed by dams dosed during gestation with 5 mg PFOA/kg (5L; lactational exposure of offspring); (3) pups exposed in utero to 5 mg PFOA/kg nursed by control dams (5U, intrauterine exposure of offspring); (4) pups exposed in utero to 5 mg PFOA/kg nursed by dams dosed during gestation with 5 mg PFOA/kg (5U + L, intrauterine and lactational exposure of offspring). Foster litters included 10 pups with equal representation of males and females (where possible). All pups were either assigned to foster litters or were euthanized.
2.5. Restricted-exposure study
Sixty-four timed pregnant CD-1 mice were received on GD 0. Mice were weighed and randomly assigned to treatment groups, and dosed orally by gavage as follows: vehicle control dosed with de-ionized water at 10 ml/kg on GD 7–17 (n = 12); 5 mg PFOA/kg on GD 7–17 (n = 14), GD 10–17 (n = 14), GD 13–17 (n = 12), or GD 15–17 (n = 12), as shown in Fig. 1C. The study did not include the GD 1–17 dosing window, as this was addressed in the 5U + L dose group in the late-life effects cross-foster study, performed within weeks of this study. On GD 18–19, mice were monitored at frequent intervals until parturition and the date and time of birth, number of live and dead pups, and number of pups of each sex were recorded and litters were weighed by sex. Litters were culled to 10 pups with equal representation of male and female (where possible).
2.6. Postnatal observations and necropsy
For the early-life effects cross-foster study, litters were observed, weighed, and euthanized on PND 1, 3, 5, and 10 (n = 4 litters per treatment group, per time point). All dams and offspring in this study were euthanized on or before PND 10. In the late-life effects cross-foster and restricted-exposure studies, litters were observed as previously described [11]. On PND 22, pups from the two latter studies were weighed and weaned, and males and females were housed separately. From these studies, female offspring were necropsied on PND 22, 29, 32, 42, and 62, as well as at 18 months postnatally. At necropsy in all studies, body and liver weight measurements were made, and whole livers and blood samples were collected. Serum was prepared from blood samples and stored frozen for PFOA analysis (due to limited volume, female pup blood was pooled by litter at necropsies which occurred prior to weaning). The fourth and fifth inguinal MG were collected from dams in the early-life effects cross-foster study and female offspring in all three studies. MG tissue from one side of the animal was prepared as a whole mount, and the contralateral glands were prepared for histological analysis.
2.7. Mammary gland preparations
MG tissues isolated at necropsy for whole mount purposes were mounted flat on glass slides. Whole mounts were then fixed in Carnoy’s solution, stained in carmine alum stain, and dehydrated and cleared in xylene, as previously described [25]. A portion of the contralateral MG was removed, placed in a histology cassette, and fixed in 10% neutral buffered formalin for 48 h, then stored in 70% ethanol. These histologically prepared glands were paraffin-embedded, and 5 μm sections were prepared and stained with hematoxylin and eosin (H&E).
Whole mounts were visualized by light macroscope (Leica WILD M420 macroscope, Leica, Wetzlar, Germany; magnification up to 70×). MG whole mounts from female offspring between PND 1 and PND 63 were scored on a 1–4 subjective, age-adjusted, developmental scale (as described in [26]; 1 = poor development/structure; 4 = normal development/structure, given age). Briefly, the developing tissue was assessed for the gross presence and appropriate timing of several histological criteria, including primary ducts and large secondary ducts, lateral side branching, appearance of budding from the ductal tree, longitudinal outgrowth of the epithelium, terminal end buds, differentiated ends, and contact inhibition between glands. Slides were separated by score as they were evaluated, compared within a score for consistency, and then recorded. Two independent scorers, blind to treatment, scored glands within the age groups. Mean scores for the time points, within treatment groups, were calculated and analyzed statistically for treatment and time-related differences.
Whole mounts from 18-month-old offspring and lactating dams were qualitatively examined with respect to concurrent controls. Areas of unusual, darkly staining foci in 18-month-old tissues were counted. Lactating glands were assessed for differentiation, amount of epithelial tissue filling the gland, and presence of well-formed, productive alveoli. Representative tissues from these assessments, as well as those scored by the above-described methods were photographed using the Leica macroscope and mounted camera (Photometrics CoolSNAP, Roper Scientific, Inc., Tucson, AZ). Histological sections were visualized by light microscope (Nikon Eclipse E600, Nikon, Tokyo, Japan), and were assessed by a pathologist for inflammation, preneoplastic lesions, and areas of hyperplasia, which might contribute to the aforementioned unusual foci. Magnifications are shown in figures. Photographs were taken using a Nikon FDX-35 scope-mounted camera.
2.8. Serum PFOA determination
Serum samples from the dams and pups of the early-life effects cross-foster study (at PND 1, 3, 5, and 10), pups of the restricted-exposure study (at PND 22, 29, and 32), and 18-month-old female offspring from the late-life effects cross-foster study were prepared at the Environmental Protection Agency. All serum samples were stored frozen in polypropylene vials, shipped on dry ice to the Centers for Disease Control and Prevention’s National Center for Environmental Health laboratory, and then kept at or below −40 °C until analysis. Measurement of the concentrations of PFOA in serum was performed through a multiple reaction monitoring experiment using a modification of the online solid-phase extraction (SPE) coupled to reversed-phase high-performance liquid chromatography (HPLC)–tandem mass spectrometry previously described [27]. A Surveyor HPLC pump (ThermoFinnigan, San Jose, CA, USA) was used, coupled with a ThermoFinnigan TSQ Quantum Ultra triple-quadrupole mass spectrometer equipped with a heated electrospray ionization (HESI) interface. The HPLC pump operated at a 300 μl/min flow rate with 20 mM ammonium acetate (pH 4) in water (mobile phase A) and acetonitrile (mobile phase B). Necessary dilution of the serum samples was performed in two steps. First, at least 10 μl serum was diluted to 0.5 ml with water in a 2-ml Eppendorf tube, then a second dilution was performed by aliquoting the appropriate amount of the diluent into an autosampler vial, adding 0.1 M formic acid, and injected into a commercial column switching system allowing for concentration of PFOA on a C18 SPE column. The column was automatically positioned in front of a Betasil C8 analytical HPLC column (2.1 mm × 50 mm, 5 μm; ThermoHypersil-Keystone, Bellefonte, PA, USA) for chromatographic identification of PFOA. Detection and quantification utilized negative-ion HESI, a variant of electrospray ionization, tandem mass spectrometry. The isotope-labeled internal standard used for quantification was 13C2 -PFOA. Blanks and quality control materials, prepared in calf serum, were analyzed along with each batch of samples to ensure the accuracy and reliability of the data across time [27].
2.9. Statistical analysis
Data were evaluated for age and exposure period effects using mixed-model analysis of variance (ANOVA) in SAS 9.1 (SAS Institute, Inc., Cary, NC). Block effects were not detected in any test and therefore block was removed from the model. For all measurements, means were evaluated and effects of dose and exposure periods compared. Statistical analysis of body weight and maternal effects for the late-life effects cross-foster study and restricted exposure study were performed as previously described [11]. In the early-life effects cross-foster study, treatment-specific mean body weights were calculated for dams at GD 1 and GD 17, and for pups, with litter as the unit of measure, on PND 1, 3, 5, and 10. For all three studies, mean MG developmental scores were calculated. MG scores were analyzed using body weight at time of collection as a random effect, with litter as the unit of measure for neonatal scores. Differences between treatment groups were determined using Dunnett’s or Tukey’s t-tests (significance at the level of p < 0.05 for all comparisons, in text and figures), with SAS.
3. Results
3.1. Late-life effects cross-foster study
As previously reported, all exposed groups exhibited lower body weight compared to controls at PND 22, except 3L [11]. These deficits were overcome within one week subsequent, except among the 5U and 5U + L groups which did not recover until as late as PND 85 [11]. Additionally, all exposed groups exhibited increased liver to body weight ratios at weaning [11], presumed to result from liver hypertrophy, as observed in adult-treated animals. In the present study, at PND 22, 42, and 63 (that is, 3-, 6- and 9-weeks-old) all cross-fostered offspring that received PFOA exposure, regardless of route or dose, exhibited reduced MG developmental scores, as compared to controls, except for the 3L group at PND 22, and the 3U + L group at PND 42 (Table 1). This abnormal development was characterized by delays in ductal elongation, delays in timing of terminal end bud (TEB) appearance, and reduced secondary and tertiary branching (Fig. 2A). Reduced branching was particularly pronounced among the combined exposure groups (3U + L, not shown; 5U + L), which exhibited mammary fat pads with greatly reduced parenchymal density (not reflected in 3U + L score at PND 42, due to inter-individual variance and the multiple criteria used to arrive at the final score). While PFOA-exposed groups at most time points exhibited lower MG developmental scores compared to controls, there was no consistent trend that suggested that any one exposure route (5L vs. 5U), or dose given via an exposure route (3L vs. 5L) negatively affected MG development more than the others.
Table 1.
Mammary gland developmental scores.
| Female offspring age in postnatal days
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| PND 1 | PND 3 | PND 5 | PND 10 | PND 22 | PND 29 | PND 32 | PND 42 | PND 63 | |
| Restricted-exposure study dose groups | |||||||||
| Control | – | – | – | – | – | 3.6 ± 0.1 | 3.6 ± 0.1 | – | – |
| GD 7–17 | – | – | – | – | – | 2.1 ± 0.1* | 1.9 ± 0.1* | – | – |
| GD 10–17 | – | – | – | – | – | 2.0 ± 0.2* | 2.2 ± 0.2* | – | – |
| GD 13–17 | – | – | – | – | – | 2.2 ± 0.1* | 2.5 ± 0.1* | – | – |
| GD 15–17 | – | – | – | – | – | 2.0 ± 0.2* | 2.3 ± 0.3* | – | – |
| Early cross-foster study dose groups | |||||||||
| Control | 3.3 ± 0.2 | 3.5 ± 0.2 | 3.0 ± 0.1 | 2.8 ± 0.2 | – | – | – | – | – |
| 5L | 1.7 ± 0.4* | 1.8 ± 0.2* | 1.5 ± 0.1* | 2.3 ± 0.2* | – | – | – | – | – |
| 5U | 1.9 ± 0.2* | 1.9 ± 0.3* | 2.0 ± 0.2* | 1.4 ± 0.1* | – | – | – | – | – |
| 5U + L | 1.5 ± 0.2* | 1.7 ± 0.3* | 1.1 ± 0.1* | 1.5 ± 0.2* | – | – | – | – | – |
| Late cross-foster study dose groups | |||||||||
| Control | – | – | – | – | 3.7 ± 0.1 | – | – | 3.2 ± 0.2 | 3.1 ± 0.2 |
| 3L | – | – | – | – | 3.0 ± 0.2 | – | – | 2.5 ± 0.2* | 2.6 ± 0.2* |
| 5L | – | – | – | – | 2.1 ± 0.2* | – | – | 2.5 ± 0.1* | 2.4 ± 0.2* |
| 3U | – | – | – | – | 1.8 ± 0.2* | – | – | 2.2 ± 0.1* | 2.6 ± 0.2* |
| 5U | – | – | – | – | 2.1 ± 0.3* | – | – | 2.2 ± 0.2* | 1.9 ± 0.2* |
| 3U + L | – | – | – | – | 1.8 ± 0.2* | – | – | 2.7 ± 0.2 | 2.6 ± 0.3* |
| 5U + L | – | – | – | – | 1.2 ± 0.2* | – | – | 1.9 ± 0.2* | 1.9 ± 0.2* |
Note. Data are presented as mean ± S.E. Scores are on a 1–4 scale; criteria adjusted for stage of development and age. Due to the outbred nature of the CD-1 mouse strain, the mean score for controls was less than 4. Dashes (–) signify time points where no measure was taken within the given study. N = 10–21 adult females per treatment group per time point for the restricted-exposure study. N = 4 litters (3 pups per litter) per treatment group per time point for the early-life effects cross-foster study. N = 9–18 adult females per treatment group per time point for the late-life effects cross-foster study. Statistical comparisons made are with controls within a given study; inter-group comparisons can be found in Section 3.
Significant treatment effect compared to control; p < 0.05.
Fig. 2.

MG development of female offspring in the late-life effects cross-foster study. (A) Whole mount preparations of mammary tissue from female offspring are shown at PND 22 (25×; a lymph node appears as a large darkly staining object), PND 42 (50×), and PND 63 (50×). Glands pictured are representative of mean respective scores (given in Table 1; N = 10–13 adult females per treatment group at PND 22, N = 9–18 per group at PND 42, N = 9–17 per group at PND 63). *Significant treatment effect by ANOVA, compared to control; p < 0.05. (B) On the left, whole mounts from representative adult female offspring at 18 months of age (16×). Large arrows indicate unusual, darkly staining foci; one small arrow in 5U indicates peripheral, localized increases in epithelial density observed in some PFOA-exposed animals at 18 months. On the right, histopathologic images from contralateral glands in the same animal show ductal areas that might account for darkly staining foci observed on whole mounts (400×; N = 5–12 females per group at 18 months). The arrow in 5U identifies an area of inflammation. In 5U + L the large arrow identifies an area of increased stromal density; the small arrow points to a potentially hyperplastic region of ductal epithelium. These ductal pathologies were observed in all treatment groups; some ductal inflammation was also seen in controls.
In female offspring from this study at 18-month postnatally, MG development could not be assessed using the scoring criteria described. However, as seen in Fig. 2B, the epithelial density within a gland appeared to be reduced in PFOA-exposed animals, particularly among the combined exposure groups (3U + L, not shown; 5U + L). Ostensibly, this arose due to a smaller starting population of parenchymal cells, and thus the subsequent reduced branching and proliferation of that ductal network resulted in sparser epithelial arborization of the fat pad. Additionally, among PFOA-exposed groups there was a tendency to exhibit higher densities of unusual, darkly staining foci in an individual gland than observed among controls (Fig. 2B; mean number of foci per gland: controls = 6.9, 3 mg/kg exposure groups = 34.3, 5 mg/kg exposure groups = 38.6). In histopathologic MG sections, as shown in Fig. 2B, these darkly staining areas appear to result from one or more of the following: hyperplasia of the ductal epithelium, infiltration by inflammatory cells into ductal regions, increases in stromal density surrounding the ducts, or inappropriate differentiation of MG ductal epithelium (Fig. 2B). These histologic analyses utilized a single section of the contralateral gland, not that used for whole mounts. As such, the observations described in histopathologic sections were not in the same tissue in which whole mount observation were made, and therefore may not explain individual darkly staining foci. In whole mounts, the composition and etiology of these foci cannot be determined, but it is presumed that histopathologic MG sections are representative, and therefore reveal what produced the appearance of these foci. In addition, peripheral, localized increases in epithelial density were visible in whole mounts of some 18-month-old PFOA-exposed offspring; however, there was not a consistent effect of dose or route of exposure on the extent of the pathology. Furthermore, these increases were strictly peripheral, and did not represent an achievement of ideal epithelial organization and content, which early time points revealed to be delayed and diminished. This study was not designed to address these histological changes in the tissue, and therefore these observations are noted, but not interpreted.
3.2. Early-life effects cross-foster study
Maternal and early neonatal indices were assessed and no effect of treatment was observed on either litter size or pup birth weight (data not shown). However, dam weight gain during pregnancy was significantly higher among dams treated with 5 mg/kg PFOA on GD 8–17 (29.2 ± 0.8 g) compared to controls (25.7 ± 1.0 g).
Among all PFOA-exposed offspring, significant delays in MG developmental morphology were observed as early as PND 1 (Fig. 3), similar to those seen at later time points in the late-life effects cross-foster study. The lactation-only exposure group nursed on treated dams and thus were exposed to PFOA for at least 12 h after parturition before the PND 1 necropsy, a period which is marked by very rapid elongation and branching of the early ductal epithelium, and may be particularly susceptible to insult by circulating PFOA. These effects persisted through the duration of the study, from PND 1 to 10, among the three exposed groups as compared to controls (Table 1). Differences in severity of developmental delays between PFOA exposure groups were only evident at PND 5 when 5U and 5U + L differed significantly from one another, and at PND 10 when 5L development, despite being worse than controls, was significantly greater than either 5U or 5U + L, which were both determined to be severe (defined as a developmental score ≤1.5; Table 1).
Fig. 3.

MG development of female offspring in the early-life effects cross-foster study. Whole mount preparations of mammary tissue from female offspring at PND 1, 3, 5 (64×), and PND 10 (50×). The arrow in 5U + L on PND 1 identifies ductal epithelium. Glands pictured are representative of mean respective scores (Table 1; N = 4 litters per treatment group at each time point; three pup glands scored per litter). *Significant treatment effect by ANOVA, compared to control; p < 0.05.
None of the PFOA-exposed groups exhibited reduced body weight compared to controls on PND 1, the age at which both intrauterine-exposed groups exhibited highest circulating PFOA levels (Table 1; Fig. 4B). While this result differs from that previously reported for the late-life effects study [11], it is important to note that gestational treatment in the early-life study did not start until GD 8, as compared to GD 1 in the late-life effects study (Fig. 1A and B). On PND 3 the combined exposure females (5U + L) weighed less than controls, and by PND 5 the other exposure groups also weighed less than controls (5L and 5U). It should be noted that MG developmental scores were consistently lower among PFOA-exposed offspring over PND 1–10, in the absence of consistent body weight disparities.
Fig. 4.

Serum PFOA concentrations in dams and female offspring from the early-life effects cross-foster study (offspring from the late-life effects study also shown on right for comparison; reported in [14]). Data are shown as mean ± S.E.M. bars; numerical values are shown for non-zero controls, and for control dams nursing treated pups at PND 1. (A) Among the dams, direct treatment with PFOA consistently yielded higher serum level than nursing treated pups (5U) alone. (B) Among offspring, lactationally exposed females (5L) exhibited serum concentrations that increased until PND 10, when they converged upon concentrations observed in the intrauterine (5U) and combined exposure (5U + L) groups. At PND 1, treated pups (5U, 5U + L) exhibited higher serum PFOA concentrations than treated dams (nursed 5L, 5U + L pups). By PND 10 all exposed offspring groups were becoming similar to one another in serum concentrations, and were becoming increasingly similar in serum concentrations to their paired dams. In the late-life effects study (right), by PND 63 serum from female offspring in all treatment groups were near background levels, at less than 1000 ng/ml. Statistical comparisons are provided in the text.
Liver to body weight ratios were elevated among intrauterine-exposed pups (5U, 5U + L) on PND 1, when their serum levels were highest (Table 2; Fig. 4B), suggesting that liver hypertrophy may be initiated prior to birth. Lactationally exposed pups also exhibited a significant elevation in this ratio by PND 5, when their circulating PFOA reached approximately 15,000 ng/ml. By this time point, all PFOA-exposed offspring exhibited increased relative liver weight, which persisted through PND 10. Considering the effect of this increased liver weight, adjusted body weights were calculated by subtracting the liver weight from the body weight of each neonate. Using this adjusted body weight as a random effect, statistics on MG scores were reevaluated and determined to be unaffected by potential growth deficits not reflected in whole body weight (data not shown).
Table 2.
Body weights and relative liver weights in dams and female offspring in the early-life effects cross-foster study.
| PND 1
|
PND 3
|
PND 5
|
PND 10
|
|||||
|---|---|---|---|---|---|---|---|---|
| Body weight (g) | Relative liver weight | Body weight (g) | Relative liver weight | Body weight (g) | Relative liver weight | Body weight (g) | Relative liver weight | |
| Dam | ||||||||
| Control, control pups | 37.4 ± 1.7 | 6.5 ± 0.4 | 39.5 ± 1.6 | 6.4 ± 0.1 | 41.8 ± 1.2 | 6.9 ± 0.2 | 45.3 ± 0.9 | 7.2 ± 0.2 |
| Control, treated (5U) pups | 33.9 ± 1.2 | 6.1 ± 0.3 | 39.1 ± 2.5 | 6.1 ± 0.1 | 38.0 ± 1.8 | 7.2 ± 0.2 | 42.6 ± 0.9 | 7.6 ± 0.2 |
| Treated, control (5L) pups | 38.2 ± 0.4 | 12.0 ± 0.4* | 37.3 ± 2.8 | 13.4 ± 0.4* | 43.7 ± 0.7 | 13.1 ± 0.2* | 46.4 ± 1.5 | 10.2 ± 0.5* |
| Treated, treated (5U + L) pups | 37.4 ± 2.1 | 12.5 ± 0.7* | 41.2 ± 1.1 | 14.1 ± 0.5* | 43.3 ± 2.2 | 12.6 ± 0.4* | 48.2 ± 1.3 | 12.9 ± 0.2* |
| Female pups | ||||||||
| Control | 1.77 ± 0.06 | 4.1 ± 0.2 | 2.29 ± 0.10 | 3.3 ± 0.1 | 3.65 ± 0.10 | 3.0 ± 0.1 | 6.07 ± 0.29 | 2.7 ± 0.02 |
| 5U pups | 1.65 ± 0.01 | 6.8 ± 0.1* | 2.07 ± 0.10 | 6.6 ± 0.1* | 2.73 ± 0.13* | 6.1 ± 0.3* | 4.51 ± 0.09* | 5.8 ± 0.1* |
| 5L pups | 1.69 ± 0.04 | 4.4 ± 0.2 | 2.25 ± 0.13 | 3.6 ± 0.2 | 3.03 ± 0.11* | 3.9 ± 0.3* | 5.64 ± 0.08* | 4.3 ± 0.1* |
| 5U + L pups | 1.60 ± 0.06 | 6.9 ± 0.2* | 1.85 ± 0.05* | 6.8 ± 0.1* | 2.43 ± 0.11* | 6.0 ± 0.1* | 4.34 ± 0.09* | 6.4 ± 0.2* |
Note. Data presented are mean ± S.E. The relative liver weight means are calculated as the average of each dam’s liver weight divided by their body weight, multiplied by 100. Pup body weight means are calculated as the average of the litter averages (of 3 female pups). As with pup body weight means, relative liver weight means were calculated by averaging the litter averages for relative liver weight. N = 4 dams or litters (N = 3 female pups per litter) per treatment group per time point for the early-life effects cross-foster study. N = 9–18 adult females per treatment group per time point for the late-life effects cross-foster study.
Significant treatment effect compared to control; p < 0.05.
Morphological examination of lactating dam MG tissue suggested profoundly altered differentiation in treated dams (nursed 5L, 5U + L pups) at PND 1, when lobulo-alveolar units appeared neither distended nor differentiated to the degree of control tissues, and a large population of adipocytes remained discernable (Fig. 5). Qualitatively, these glands appeared immature, similar to that seen in late pregnancy, prior to parturition and the initiation of nursing. Because this phenotype was present in dams nursing controls pups, it may be presumed that this effect is a direct action of PFOA on the differentiating lactating dam, rather than a result of possible poor suckling and stimulation by smaller, and presumably weaker, PFOA-treated pups (as discussed in [17]). While these treated groups continued to exhibit diminished lactational morphology until PND 10, alveolar units filled the fat pad more completely by PND 3 though they did not catch up with control dam morphology before the end of the study (data not shown). Reduced alveolar filling of the fat pad was visible among control dams nursing treated offspring (5U) as early as PND 3 (data not shown), at which time point these dams exhibited serum PFOA levels of roughly 2000 ng/ml (Fig. 4A), presumably as a result of dam behaviors that include grooming the litter and stimulating micturition, thereby ingesting PFOA eliminated from the offspring. The precipitous drop in circulating PFOA in 5U pups suggests that the compound, primarily lost through the urine, is being excreted at a high rate, and could therefore readily be ingested through normal maternal behaviors. Delays in lactational morphology persisted to PND 10 among these three exposed groups of dams, with combined-exposure dams in the 5U + L group exhibiting the most profound delays (data not shown).
Fig. 5.
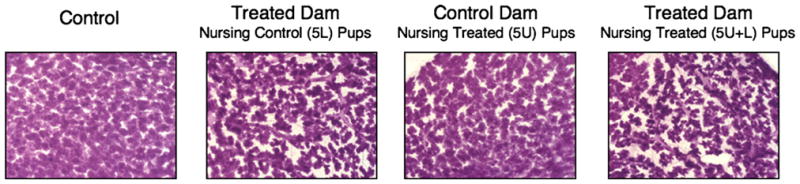
MG differentiation of lactating dams in the early-life effects cross-foster study. Whole mount preparations of mammary tissue from lactating dams are shown on PND 1, the first day of lactation (40×). Glands pictured are representative of lactating dams in respective groups at LD 1 (body weights given in Table 2; N = 4 dams per treatment group at each time point).
3.3. Restricted-exposure study
In the restricted-exposure study, treatment with PFOA under even the shortest duration treatment, GD 15–17, was sufficient to consistently lower MG scores at PND 29 and 32 as compared to controls (Table 1). These delays, observed in all treatment groups, were similar to those observed in both cross-foster studies, in that they were marked by reduced ductal elongation and branching, as well as delays in timing and density of TEBs (control, GD 15–17, and GD 10–17 shown, Fig. 6A). Because all treated groups except for GD 10–17, recovered from body weight deficits within 1 week of weaning (previously reported in [11]), the observed MG delays again occurred in the absence of body weight deficits, which might otherwise have been a factor in MG growth impairment (except in the GD 10–17 group, when body weight was still below that of controls).
Fig. 6.
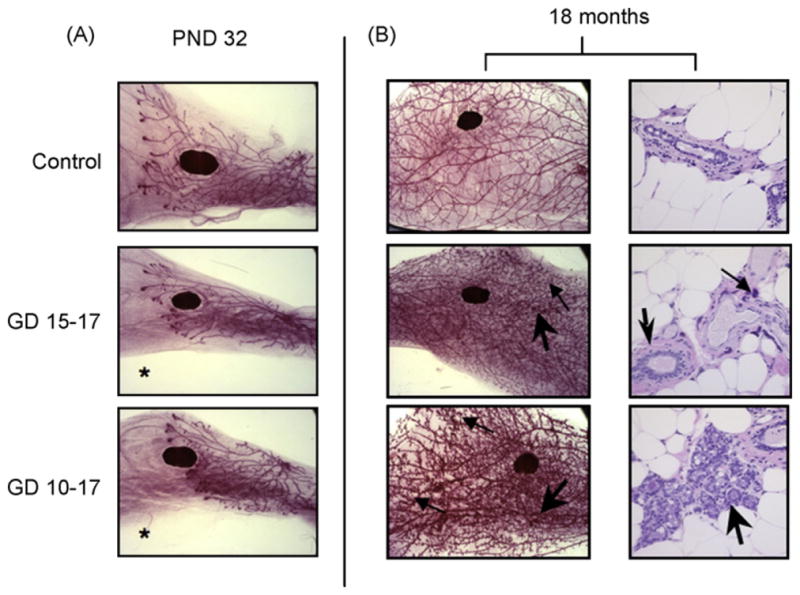
MG development of female offspring in the restricted-exposure study. (A) Whole mount preparations of mammary tissue are shown at PND 32 (16×). Glands pictured show morphology representative of respective treatment groups at given time points (Table 1; N = 10–20 females per treatment group at PND 32). *Significant treatment effect by ANOVA, compared to control; p < 0.05. (B) On the left, whole mount preparations of mammary tissue at 18 months of age are shown (16×). Large arrows identify unusual, darkly staining foci; small arrows identify peripheral, localized increases in epithelial density observed in some PFOA-exposed animals at 18 months. On the right, histopathologic images from contralateral glands in the same animal show ductal areas that might account for darkly staining foci observed on whole mounts (400×; N = 4–11 females per treatment group at 18 months). The large arrow in GD 15–17 identifies an area of increased stromal density; the small arrow points to a focus of inflammation. In GD 10–17 the arrow identifies a large, potentially hyperplastic region of ductal epithelium. These ductal pathologies were observed in all treatment groups; some ductal inflammation was also seen in controls.
At 18-month postnatally, MG development was not scored in the fully mature gland, due to the absence of developmental indices integral to scoring criteria. However, among PFOA-exposed groups, as with the late-life cross-foster study, there was a tendency for PFOA-exposed females to exhibit higher densities of unusual darkly staining foci, which were originally suspected to be areas of ductal hyperplasia (Fig. 6B; mean number of foci per gland: controls = 1.5, GD 15–17 exposure = 29.8, GD 13–17 exposure = 17.9, GD 10–17 = 32.8, GD 7–17 = 25.5). Identical to that noted in the late-life effects cross-foster study, observations in histopathologic MG sections (Fig. 6B) suggest these darkly staining foci result from one or more of the following: hyperplasia of the ductal epithelium, infiltration by inflammatory cells into ductal regions, increases in stromal density surrounding the ducts, or inappropriate differentiation of MG ductal epithelium. To a greater degree than the late-life effects study, peripheral, localized increases in epithelial density were visible in whole mounts from many 18-month-old PFOA-exposed animals.
Serum PFOA levels were not measured in these animals, though the serum data from the late-life effects cross-foster study at 18-month demonstrate that serum PFOA are at background levels. Mean body weights, liver weights, and liver to body weight ratios were similar between all treatment groups and controls at 18-month postnatally.
3.4. Serum PFOA dosimetry
The serum PFOA concentrations previously reported by Wolf et al. [11] for the late-life effects cross-foster study should be considered when evaluating the MG scores. Therefore, the control, 5L, 5U, and 5U + L group means at 3, 6, and 9 weeks [11] are presented in Fig. 4B (3L, 3U, and 3U + L are not shown, but were consistently lower than the 5 mg/kg equivalent exposure groups). These data appear alongside serum PFOA concentrations measured in the early-life effects cross-foster study (vide infra) to illustrate the potential pattern of PFOA serum load over time, based on exposure parameters. However, the early-life effects study exposure period was about 60% the duration of the late-life study, and therefore these data cannot be directly compared. Most importantly, in the late-life effects study the persistent nature of the MG developmental delays was evident at 9 weeks postnatally, when the circulating PFOA concentrations had returned to near-background levels, on the order of hundreds rather than tens of thousands of ng/ml. As presumed, based on the 16–19 days half-life of PFOA in mice [28], mean serum PFOA concentrations were similar to controls and had reached background levels in all PFOA-exposed females, 18 months after their last treatment (data not shown).
In the early-life effects cross-foster study, serum PFOA concentrations in female offspring were highest at the earliest time point assessed, PND 1, among those with intrauterine exposure (5U, 5U + L; Fig. 4B). Conversely, among the lactationally exposed (5L) offspring, levels rose steadily after birth and through PND 10, demonstrating substantial postnatal lactational transfer of PFOA. This transfer was evident also by the decline in serum PFOA observed in the treated dams that nursed these pups, which exhibited the highest serum concentrations at PND 1, falling steadily during lactation (Fig. 4A). Interestingly, at PND 1 the intrauterine-exposed offspring (5U, 5U + L) exhibited significantly higher serum levels than treated dams, a surprising observation given previous assumptions about gestational transfer [29]. This trend appeared to diminish with time, and by PND 10 dam and pup serum levels were similar. Among lactating dams in this study, serum PFOA concentrations decreased over time between PND 1–10, among the two treated groups. Control dams that nursed intrauterine-only exposed pups exhibited increasing serum PFOA over the course of the study, and significantly higher serum PFOA concentrations than controls as early as PND 1, which persisted through the study.
In the restricted-exposure study, serum PFOA concentrations for all treated groups at PND 29 and 32 (previously reported in [11]) were consistently higher than controls even under the shortest duration exposure of GD 15–17. While serum PFOA concentrations were not measured in 18-month-old females from this study, one may presume – given PFOA pharmacokinetics in the CD-1 mouse [28] – that the serum concentrations for the 18-month-old 5U + L females in the late-life effects study represent conservative estimates for circulating PFOA concentrations in females of the same age, strain, and environmental conditions in the restricted-exposure study, where PFOA dose and route of exposure were the same, only exposure periods were shorter (Fig. 1A and C). Furthermore, serum PFOA reported previously for these two studies [11] consistently showed lower serum PFOA concentrations among all PFOA-treated groups in the restricted-exposure study, as compared to 5U + L offspring in the late-life effects cross-foster study at PND 22.
4. Discussion
These studies have demonstrated the capacity for both short duration prenatal and exclusively postnatal, lactational PFOA exposure to delay development of the proliferating MG in offspring from as early as PND 1, to as late as or later than 9 weeks postnatally. Furthermore, these delays remain apparent even as the internal PFOA dose drops, approaching background levels. These data, in conjunction with sparse epithelial filling of the MG fat pad observed in 18-month-old PFOA-exposed offspring, suggest that early life exposure may result in permanent effects in the mammary tissue.
In the late-life effects study, lactational-only exposure at 5 mg/kg was sufficient to produce delays in MG development that persisted from 3 weeks until at least 9 weeks of age, when sexual maturity has been attained, even though PFOA serum concentrations were relatively low (<350 ng/ml at 9 weeks), and of the same order of magnitude as concentrations reported in humans exposed to PFOA occupationally or accidentally, by ingestion of contaminated drinking water [12]. In this group, body weight deficits were overcome after PND 22 [11], yet MG developmental deficits persisted, and appeared evident even at 18 months postnatally. These effects were observed in this group despite receiving exclusively postnatal exposure. The 3/5U and 3/5U + L groups also exhibited these delays, and although body weight deficits among these groups persisted longer, serum PFOA concentrations in 3/5U females were less than or equal to that in 3/5L females for the entire 3–9 weeks of age window [11]. Even at 18 months postnatally, filling of the gland by epithelium was visibly different in density and organization, from controls in lactational exposure-only females, as well as the intrauterine exposure groups.
The early-life effects cross-foster study supported these findings, and all PFOA-exposed female offspring – 5 L, 5U, and 5U + L –exhibited delayed MG development as early as PND 1, or at least 12 h after parturition, during which time the MG parenchyma normally undergoes rapid growth and development [30]. These delays were maintained, and persisted through PND 10. These studies taken together suggest that MG deficits resulting from intrauterine, lactational, or combined exposures to PFOA develop at least as early as PND 1, and persist into sexual maturation. The observations in the early-life effects study also specifically point to a window of sensitivity for the MG during late fetal and early neonatal life. Interestingly, among the 3 mg/kg lactationally exposed group in the late-life effects study, body weight was never reduced compared to controls, yet MG morphology revealed growth deficits at PND 42 and PND 63 in the absence of any body growth retardation over life. This suggests that the threshold for effect may be lower for MG developmental delays as compared to that for body growth deficits, and that the mechanisms responsible for these effects may differ.
In the restricted-exposure study, similar persistence of MG morphological deficits was observed, with as short a treatment window as the final 3 days of gestation (GD 15–17) producing delays visible at PND 29 and PND 32, and persistent morphological changes visible at 18 months. These findings are consistent with those of the cross-foster studies, demonstrating the late gestation and early lactation periods as most sensitive for the effects of PFOA on the MG.
These findings support previous work [17] that reported delays in MG development resulting from developmental PFOA exposure, independent of body weight deficits. This work is also in agreement with work on other environmental agents, which indicates that late fetal and early neonatal MG development is particularly sensitive to environmental insult (reviewed in [31], where the rodent MG is illustrated to be a suitable model for the human tissue). Furthermore, this is the first work to the authors’ knowledge that reports effects of developmental PFOA exposure occurring as late as the postnatal period – via the presumed lower transmission route of nursing – that persist into adulthood and late-life. An important finding was also illuminated in the dosimetry data, specifically that offspring with intrauterine PFOA exposure exhibited higher serum PFOA concentrations on PND 1 than did the treated dams they nursed on, an observation not previously reported, and vital to the understanding of the pharmacokinetics of PFOA in the fetus and neonate. It deserves mention that the reduced MG epithelium phenotype reported here was also recently observed following postnatal, peripubertal exposure of C57BL/6 (at the dose of 10 mg/kg, only) and Balb/c mice to PFOA [32, this issue]. The finding of this similar study, that the response in the MG seems to differ between inbred strains, is noteworthy and merits further investigation.
At the conclusion of these studies, numerous questions remain unanswered. Understanding the no observable adverse effect level (NOAEL) and the lowest observable adverse effect level (LOAEL) in conjunction with internal dosimetry would be beneficial in reducing uncertainties in the relationship between dose and health effects. Using the data presented here and elsewhere, pharmacokinetic models may be able to anticipate these doses. Characterizing the long-term adverse effects of MG perturbations in PFOA exposed animals, including possible impaired lactational support of litters or altered cancer susceptibility also remains to be understood. Finally, how these morphological delays and subsequent persistent deficits are mediated remains unclear, but will be important in determining the human health hazard posed by PFOA.
PFOA is a known agonist of the peroxisome proliferator-activated receptor (PPAR) α isoform (PPARα). Knockout and transgenic mice have provided some clues as to possible modes of action for PFOA in the MG. In transgenic mouse dams exhibiting constituitively activated PPARα, normal differentiation of lobular–alveolar units was so profoundly impaired during lactation, that no offspring survived until weaning [33], suggesting that altered PPARα-signalling has the potential to greatly interfere with proper functional differentiation of the lactating gland.
The mode of action responsible for the general growth deficits observed in gestationally PFOA-exposed mice was recently examined using PPARα knockout mice. These effects – including impaired postnatal body weight gain, delayed eye opening among pups, and postnatal mortality – were found to be dependent upon PPARα expression [16]. Interestingly, early prenatal loss did not appear to require PPARα expression. This study did not examine dam lactation or MG morphology of female offspring, however, studies addressing these tissues in PPARα knockout mice are currently underway. Of note is the fact that the PPARα knockout mice do seem to reproduce normally and exhibit normally developed MG tissue. It is noteworthy that MG effects have been observed to occur in the absence of growth deficits, which have been identified as PPARα-dependent effects. Furthermore, because these delays in body weight gain and developmental indices were shown not to result from lactational exposure only, while MG effects were, there is evidence that MG developmental effects may not be mediated by a similar mechanism and may represent a more sensitive endpoint.
While the observations reported here and in previous publications [17] concerning impaired lactational development of lobular–alveolar units among PFOA-treated dams, agree with similar observations in publications examining the impact of PPARα agonists on lactation [33], the authors do not rule out the potential for an upstream event or an entirely PPARα-independent pathway to be responsible for the MG effects in either the treated dam or the exposed offspring.
This work has identified the MG as a tissue that is sensitive to developmental perturbation by PFOA, and may be among the most sensitive endpoints studied to date. In these studies, when internal dose was examined with respect to effect, a circulating serum concentration of about 2000 ng/ml appeared sufficient to stimulate the inhibition of developing and differentiating MG tissue. This is approximately four times the circulating PFOA concentration found in some non-occupationally exposed populations [12], suggesting that epidemiologic studies focused on the health effects of PFOA exposure may want to evaluate women’s ability to exclusively breast feed. With respect to animal studies, knowledge of the lowest effective doses and the mode of action for the PFOA-induced MG effects will allow comparison with health results in epidemiological studies that are currently on-going, including those by the Breast Cancer and Environment Research Centers [34] and by the C8 Science Panel [35] on the human health effects of PFOA. This type of research, in conjunction with internal dosimetry, should result in less uncertainty in risk assessment.
Acknowledgments
The authors thank Dr. David Kurtz and the technical staff at New Year Tech, Inc. for their exceptional animal care during this lengthy study. This study was supported by the U.S. Environmental Protection Agency (EPA). Financial support for SS White provided by the U.S. EPA, NHEERL-UNC Toxicology Curriculum Cooperative Training Agreements (T 829472 and CR 833237) with the University of North Carolina, Chapel Hill, NC, 27599.
Footnotes
The information in this document has been funded by the U.S. Environmental Protection Agency. It has been subjected to review by the National Health and Environmental Effects Research Laboratory and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use. Furthermore, the findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control. Portions of these data within this article were presented at the Society of Toxicology Meeting in Charlotte, NC, March 2007.
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- 1.Giesy JP, Kannan K. Perfluorochemical surfactants in the environment. Environ Sci Technol. 2002;36:146A–52A. doi: 10.1021/es022253t. [DOI] [PubMed] [Google Scholar]
- 2.Hansen KJ, Johnson HO, Eldridge JS, Butenhoff JL, Dick LA. Quantitative characterization of trace levels of PFOS and PFOA in the Tennessee River. Environ Sci Technol. 2002;36:1681–5. doi: 10.1021/es010780r. [DOI] [PubMed] [Google Scholar]
- 3.Harada K, Saito N, Inoue K, Yoshinaga T, Watanabe T, Sasaki S, et al. The influence of time, sex and geographic factors on levels of perfluorooctane sulfonate and perfluorooctanoate in human serum over the last 25 years. J Occup Health. 2004;46:141–7. doi: 10.1539/joh.46.141. [DOI] [PubMed] [Google Scholar]
- 4.Hoff PT, Van de Vijver K, Van Dongen W, Esmans EL, Blust R, De Coen WM. Perfluorooctane sulfonic acid in bib (Trisopterus luscus) and plaice (Pleuronectes platessa) from the Western Scheldt and the Belgian North Sea: distribution and biochemical effects. Environ Toxicol Chem. 2003;22:608–14. [PubMed] [Google Scholar]
- 5.Kannan K, Corsolini S, Falandysz J, Oehme G, Focardi S, Giesy JP. Perfluorooctanesulfonate and related fluorinated hydrocarbons in marine mammals, fishes, and birds from coasts of the Baltic and the Mediterranean Seas. Environ Sci Technol. 2002;36:3210–6. doi: 10.1021/es020519q. [DOI] [PubMed] [Google Scholar]
- 6.Key BD, Howell RD, Criddle CS. Fluorinated organics in the biosphere. Environ Sci Technol. 1997;31:2445–54. [Google Scholar]
- 7.Kubwabo C, Vais N, Benoit FM. A pilot study on the determination of perfluorooctanesulfonate and other perfluorinated compounds in blood of Canadians. J Environ Monit. 2004;6:540–5. doi: 10.1039/b314085g. [DOI] [PubMed] [Google Scholar]
- 8.Olsen GW, Burris JM, Burlew MM, Mandel JH. Epidemiologic assessment of worker serum perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations and medical surveillance examinations. J Occup Environ Med. 2003;45:260–70. doi: 10.1097/01.jom.0000052958.59271.10. [DOI] [PubMed] [Google Scholar]
- 9.Olsen GW, Church TR, Miller JP, Burris JM, Hansen KJ, Lundberg JK, et al. Perfluorooctanesulfonate and other fluorochemicals in the serum of American Red Cross adult blood donors. Environ Health Perspect. 2003;111:1892–901. doi: 10.1289/ehp.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ Health Perspect. 2007;115:1596–602. doi: 10.1289/ehp.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf CJ, Fenton SE, Schmid JE, Calafat AM, Kuklenyik Z, Bryant XA, et al. Developmental toxicity of perfluorooctanoic acid in the CD-1 mouse after cross-foster and restricted gestational exposures. Toxicol Sci. 2007;95:462–73. doi: 10.1093/toxsci/kfl159. [DOI] [PubMed] [Google Scholar]
- 12.Emmett EA, Shofer FS, Zhang H, Freeman D, Desai C, Shaw LM. Community exposure to perfluorooctanoate: relationships between serum concentrations and exposure sources. J Occup Environ Med. 2006;48:759–70. doi: 10.1097/01.jom.0000232486.07658.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy GL, Jr, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, et al. The toxicology of perfluorooctanoate. Crit Rev Toxicol. 2004;34:351–84. doi: 10.1080/10408440490464705. [DOI] [PubMed] [Google Scholar]
- 14.U.S. Environmental Protection Agency [EPA], Office of Pollution Prevention and Toxics, Risk Assessment Division. SAB review of EPA’s draft risk assessment of the potential human health effects associated with exposure to perflurooctanoic acid and its salts (EPA-SAB-06-006) Washington, DC: Science Advisory Board Perfluorooctanoic Acid Review Panel; 2006. [Google Scholar]
- 15.Lau C, Thibodeaux JR, Hanson RG, Narotsky MG, Rogers JM, Lindstrom AB, et al. Effects of perfluorooctanoic acid exposure during pregnancy in the mouse. Toxicol Sci. 2006;90:510–8. doi: 10.1093/toxsci/kfj105. [DOI] [PubMed] [Google Scholar]
- 16.Abbott BD, Wolf CJ, Schmid JE, Das KP, Zehr RD, Helfant L, et al. Perfluo rooctanoic acid induced developmental toxicity in the mouse is dependent on expression of peroxisome proliferator activated receptor-alpha. Toxicol Sci. 2007;98:571–81. doi: 10.1093/toxsci/kfm110. [DOI] [PubMed] [Google Scholar]
- 17.White SS, Calafat AM, Kuklenyik Z, Villanueva L, Zehr RD, Helfant L, et al. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci. 2007;96:133–44. doi: 10.1093/toxsci/kfl177. [DOI] [PubMed] [Google Scholar]
- 18.Apelberg BJ, Witter FR, Herbstman JB, Calafat AM, Halden RU, Needham LL, et al. Cord serum concentrations of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in relation to weight and size at birth. Environ Health Perspect. 2007;115:1670–6. doi: 10.1289/ehp.10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fei C, McLaughlin JK, Tarone RE, Olsen J. Perfluorinated chemicals and fetal growth: a study within the Danish National Birth Cohort. Environ Health Perspect. 2007;115:1677–82. doi: 10.1289/ehp.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fei C, McLaughlin JK, Tarone RE, Olsen J. Fetal growth indicators and perfluorinated chemicals: a study in the Danish National Birth Cohort. Am J Epidemiol. 2008;168:66–72. doi: 10.1093/aje/kwn095. [DOI] [PubMed] [Google Scholar]
- 21.Kärrman A, Ericson I, van Bavel B, Darnerud PO, Aune M, Glynn A, et al. Exposure of perfluorinated chemicals through lactation: levels of matched human milk and serum and a temporal trend, 1996–2004, in Sweden. Environ Health Perspect. 2007;115:226–30. doi: 10.1289/ehp.9491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monroy R, Morrison K, Teo K, Atkinson S, Kubwabo C, Stewart B, et al. Serum levels of perfluoroalkyl compounds in human maternal and umbilical cord blood samples. Environ Res. 2008 doi: 10.1016/j.envres.2008.06.001. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 23.Tao L, Kannan K, Wong CM, Arcaro KF, Butenhoff JL. Perfluorinated compounds in human milk from Massachusetts, U.S.A. Environ Sci Technol. 2008;42:3096–101. doi: 10.1021/es702789k. [DOI] [PubMed] [Google Scholar]
- 24.Völkel W, Genzel-Boroviczény O, Demmelmair H, Gebauer C, Koletzko B, Twardella D, et al. Perfluorooctane sulphonate (PFOS) and perfluorooctanoic acid (PFOA) in human breast milk: results of a pilot study. Int J Hyg Environ Health. 2008;211:440–6. doi: 10.1016/j.ijheh.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 25.Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2002;67:63–74. doi: 10.1093/toxsci/67.1.63. [DOI] [PubMed] [Google Scholar]
- 26.Rayner JL, Wood C, Fenton SE. Exposure parameters necessary for delayed puberty and mammary gland development in Long-Evans rats exposed in utero to atrazine. Toxicol Appl Pharmacol. 2004;195:23–34. doi: 10.1016/j.taap.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Kuklenyik Z, Needham LL, Calafat AM. Measurement of 18 perfluorinated organic acids and amides in human serum using on-line solid-phase extraction. Anal Chem. 2005;77:6085–91. doi: 10.1021/ac050671l. [DOI] [PubMed] [Google Scholar]
- 28.Lau C, Strynar MJ, Lindstrom AB, Hanson RG, Thibodeaux JR, Barton HA. Pharmacokinetic evaluation of perfluorooctanoic acid in the mouse. Toxicologist. 2005;84:252. [Google Scholar]
- 29.Hinderliter PM, Mylchreest E, Gannon SA, Butenhoff JL, Kennedy GL., Jr Perfluorooctanoate: placental and lactational transport pharmacokinetics in rats. Toxicology. 2005;211:139–48. doi: 10.1016/j.tox.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Daniel CW, Silberstein GB. Postnatal development of the rodent mammary gland. In: Neville MC, Daniel CW, editors. The mammary gland: development, regulation, and function. New York: Plenum Press; 1987. pp. 3–36. [Google Scholar]
- 31.Fenton SE. Endocrine-disrupting compounds and mammary gland development: early exposure and later life consequences. Endocrinology. 2006;147:S18–24. doi: 10.1210/en.2005-1131. [DOI] [PubMed] [Google Scholar]
- 32.Yang C, Tan YS, Harkema JR, Haslam SZ. Differential effects of peripubertal exposure to perfluorooctanoic acid on mammary gland development in C57Bl/6 and Balb/c mouse strains. Reprod Toxicol. doi: 10.1016/j.reprotox.2008.10.003. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Q, Kurotani R, Yamada A, Kimura S, Gonzalez FJ. PPARα activation during pregnancy severely impairs mammary lobuloalveolar development in mice. Endocrinology. 2006;147:4772–80. doi: 10.1210/en.2006-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. [Accessed August 8, 2008];Breast Cancer and Environment Research Centers. 2008 Available at: http://www.bcerc.org.
- 35. [Accessed August 8, 2008];C8 Science Panel. 2008 Available at: http://www.c8sciencepanel.org.