

Abstract
TOR (target of rapamycin) is an evolutionarily conserved nutrient sensing protein kinase that regulates growth and metabolism in all eukaryotic cells. Studies in flies, worms, yeast and mice support the notion that the TOR signaling network plays a pivotal role in modulating aging. TOR is emerging as the most robust mediator of the protective effects of various forms of dietary restriction (DR), which has been shown to extend lifespan and slow the onset of certain age-related diseases across species. Here we discuss how modulating the TOR signaling network slows aging by affecting a number of downstream processes including mRNA translation, autophagy, endoplasmic reticulum (ER) stress signaling, stress responses and metabolism. Identifying the mechanisms by which the TOR signaling network works as a pacemaker of aging is a major challenge in the field and may help identify potential drug targets for age–related diseases thereby facilitating healthful lifespan extension in humans.
Introduction
TOR belongs to a conserved group of serine/threonine kinases from the phosphatidylinositol kinase-related kinase (PIKK) family. Rapamycin, an immunosuppressive macrolide, that was first discovered as the product of the bacterium Streptomyces hygroscopicus from the Easter Island (Rapa Nui), inhibits the activity of TOR (Heitman et al., 1991). TOR, first identified in yeast and since then in all eukaryotes examined, exists in two distinct complexes with different functions (Martin and Hall, 2005). TOR complex I (TORC1) is rapamycin sensitive and is the central element of the TOR signaling network (Figure 1). It monitors and integrates a diverse set of intra- and extracellular parameters and controls cell size, proliferation and lifespan via a variety of downstream pathways. It is composed of the Serine/Threonine kinase TOR, and its associated proteins Raptor (regulatory associated protein of TOR), mLst8, PRAS40, and, in mammals, Deptor (Guertin and Sabatini, 2005, 2009; Wullschleger et al., 2006). On the other hand, TOR complex 2 (TORC2) is rapamycin-insensitive and controls the activity of Serum and Glucocorticoid-induced kinase (SGK) and contributes to the full activation of Akt (Alessi et al., 2009). It contains TOR, Rictor (rapamycin-insensitive companion of mTOR), Sin1, Proctor/PRR5L, mLst8 and (again, only in mammals) Deptor (Guertin and Sabatini, 2005, 2009; Wullschleger et al., 2006).
Figure 1. Schematic of the TOR signaling network.
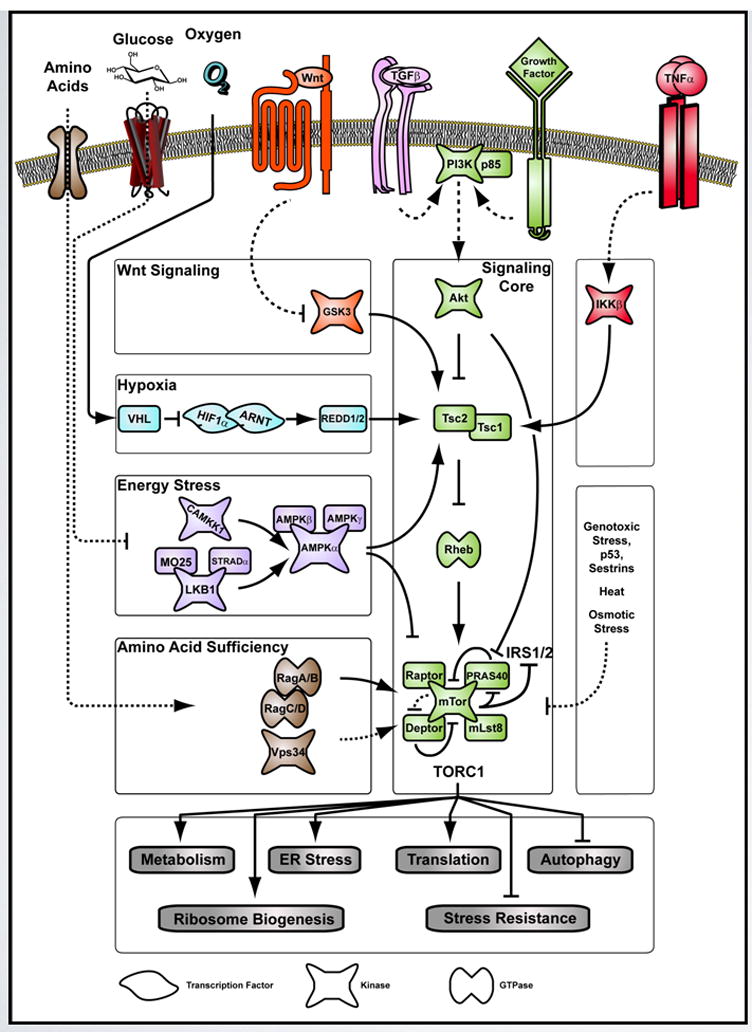
TORC1 integrates intra- and extracellular environmental cues through discrete signaling modules that sense and relay diverse inputs to a central “signaling core” (in green). This figure summarizes biochemical evidence from various studies that have identified the inputs and outputs of TORC1. Various outputs (in grey) that keep cellular growth in balance with the environment are regulated by TORC1. See main text for details.
The TOR signaling network can be schematized by connecting discrete signaling modules sensing and relaying diverse inputs to a central “signaling core” (Figure1). This core consists of the Serine/Threonine kinase Akt, the Tuberous Sclerosis Complex proteins Tsc1 and Tsc2, the Ras-like small GTPase Rheb and TORC1 itself. In mammals the activity of TORC1 is held in check by three inhibitory elements within this cascade: the Tsc1/Tsc2 complex, PRAS40 and Deptor (Peterson et al., 2009). The biochemical nature of how the signal cascades though the pathway has been exquisitely reviewed and is only summarized here (Guertin and Sabatini, 2009; Shamji et al., 2003; Shaw and Cantley, 2006; Wullschleger et al., 2006). In short, the activation of the TORC1 signaling core occurs by an Akt-driven de-repression mechanism. Once activated by upstream signals, Akt phosphorylates and inhibits the negative regulators Tsc2 and PRAS40, which then activates the GTPase Rheb and releases TORC1 from inhibition.
Despite the evolutionary conservation of individual components of the TOR pathway, some significant differences exist between species. Of note, baker’s yeast (S. cerevisiae) and fission yeast (S. pombe) contain two genes encoding TOR, which are differentially incorporated into the distinct TORC complexes (Loewith et al., 2002). However, S. pombe hosts genes for the Rheb GTPase and the Tsc1/Tsc2 complex, while S. cerevisiae does not. The nematode C. elegans lacks a functional equivalent to the Tsc1/Tsc2 complex, but connects the activity of Akt and TORC1 by Akt dependent transcriptional regulation of Raptor (Jia et al., 2004; Wullschleger et al., 2006). Despite these differences, the TOR signaling network controls a conserved set of physiological processes in multiple species (see below).
TOR plays a conserved role in coupling nutrients to growth
Since its discovery as a regulator of growth in S. cerevisiae, TOR has been shown to be a conserved nutrient sensor across species. This has been reflected in the common loss-of-function phenotypes of TOR in multiple species. In C. elegans, deletion of CeTOR leads to developmental arrest at the L3 larval stage and intestinal atrophy (Long et al., 2002). A similar phenotype was observed for mutants in daf-15, the worm ortholog of the mammalian protein raptor (Jia et al., 2004). In D. melanogaster, larvae lacking TOR show similarities to amino acid-deprived animals, such as developmental arrest, reduced nucleolar size and lipid vesicle aggregation in the larval fat body (Oldham et al., 2000; Zhang et al., 2000). Similar to invertebrates, loss of function of mammalian TOR (mTOR) in mice leads to early embryonic lethality (Hentges et al., 2001; Murakami et al., 2004). The tissue-specific removal of raptor in skeletal muscle induces progressive muscle dystrophy, which is likely due to impaired protein synthesis (Bentzinger et al., 2008). Thus, mutations in TOR show developmental or growth arrest phenotypes in different species, comparable to those observed upon nutrient deprivation, supporting the notion that it has a conserved role in coupling of availability of nutrients to growth in multiple species.
TOR integrates extra- and intracellular signals from nutrients, growth factors and various stresses
In addition to playing a conserved role as a common nutrient sensor, TORC1 acts as a major hub integrating signals emanating from a variety of sources and influencing multiple physiological responses (Figure 1). TORC1 receives signals from growth factors, nutrients, energy status and a range of stressors. This has lead to the view that TOR signaling serves as a ‘growth checkpoint,’ surveying the extra and intracellular milieu for favorable conditions to mediate growth accordingly (Shaw and Cantley, 2006). This ability to balance a multitude of signals is reflected in the number of signaling modules linked to the central signaling core (Figure 1). Multiple inputs are integrated and relayed towards the regulation of a number of downstream pathways. TORC1 activity influences mRNA translation, autophagy, transcription, metabolism and cell survival, proliferation, size and growth, in addition to number of other cellular processes. An accumulating body of data suggests that some of these responses influence organismal lifespan.
To ensure that growth rates match resources, both the quality and quantity of the nutrient status needs to be assessed and integrated. It seems that the eukaryotic cell has solved this problem by utilizing the TOR signaling network as an important checkpoint (Figure 1). Withdrawal of amino acids acts as a powerful inhibitor of TORC1 activity, even in the presence of acute growth factor stimulation (Hara et al., 1998). Essential amino acids like L-tryptophan, L-phenylalanine, L-arginine and especially L-leucine activate TORC1 after being transported into the cell in an L-glutamine-dependent manner (Nicklin et al., 2009). While it remains unclear how amino acid sufficiency is sensed within the cytoplasm of the cell, two small heterodimeric GTPases, RagA/RagC and RagB/RagD have been shown to relay the signal of amino acid sufficiency to TORC1 (Kim et al., 2008; Sancak et al., 2008). TOR also responds to the energy charge within the cell which is influenced by the presence of nutrients like glucose. A drop in the cell’s energy content is reflected in the rise of the AMP/ATP ratio and triggers AMP-dependent activation of AMP dependent kinase (AMPK) (Hardie, 2007). In turn, AMPK reduces the activity of the TOR signaling core by direct phosphorylation on two key proteins: It phosphorylates and stimulates Tsc2 activity, and further reduces TORC1 by inhibitory Raptor phosphorylation (Gwinn et al., 2008). In addition to nitrogen starvation, carbon and phosphate starvation also lead to inhibition of TORC1 in S. cerevisiae (Kuruvilla et al., 2001; Shamji et al., 2000; Urban et al., 2007). Hence, the TOR pathway integrates information on cellular status by sensing both qualitative and quantitative changes in nutrients.
As noted above, unicellular organisms sense and directly respond to the presence or absence of nutrients, such as amino acids and glucose, to control growth. In multicellular organisms, the requirement for coordination of growth in different tissues creates a need for intercellular communication, which is achieved by diffusible growth factors. In order to gauge the appropriateness of these growth signals, multicellular organisms have evolved to integrate various growth factors and direct nutrient inputs. The role of TORC1 as a major hub for integration of control of growth is supported by the observations that its activity is modulated by various growth-signaling pathways. Growth factors like IGF and insulin modulate TORC1 activity through Akt. Wnt signaling has also been demonstrated to regulate TORC1 activity (Inoki and Guan, 2006). Stimulation of the Wnt receptors culminates in the inhibition of glycogen synthase kinase 3 (Gsk3). When active, Gsk3 has been shown to directly phosphorylate and activate Tsc2 when primed by AMPK-dependent phosphorylation (Inoki et al., 2006). Therefore, Wnt mediated inactivation of Gsk3 alleviates Tsc2-driven inhibition of Rheb and results in TORC1 activation. Inputs from various other growth factors, like that from transforming growth factor (TGF) beta (Lamouille and Derynck, 2007), further endorse TORC1’s role in sensing extracellular growth status.
In addition to nutrients and growth factors, appropriate control of cell growth also requires integration of information on environmental stresses. Hence, it is not surprising that multiple stresses also modulate TORC1 activity. TORC1 is rendered less active by high temperature, hydrogen peroxide and high salt stress in S. cerevisiae (Urban et al., 2007). Correspondingly, osmotic stress inactivates TORC1 by modulating mitochondrial function (Desai et al., 2002). TORC1 also responds to hypoxia. Low oxygen levels stabilize HIF1α, allowing the formation of a heterodimer with the aryl hydrocarbon receptor nuclear translocator (ARNT). The HIF1a/ARNT dimer activates the transcription of hypoxic response genes, most notably REDD1 that inhibits TORC1 activity by a TSC2-dependent mechanism (Brugarolas et al., 2004; Reiling and Hafen, 2004). Genotoxic stress has also been shown to inhibit TORC1. Sestrin1 and Sestrin2, both transcriptional targets of the DNA damage sensor p53, activate AMPK, thereby inhibiting TOR pathway activity (Budanov and Karin, 2008). Hence, sensing of inputs from nutrients, growth factors and cellular stress signals, uniquely positions TORC1 to synchronize growth in tune with its environment.
In summary, the TOR signaling network integrates information from a variety of inhibitory and stimulatory environmental signals. Some of the key questions in the field include: 1) how are multiple nutrients, growth factors and stress inputs quantitatively integrated into a single TORC1 activity level, 2) are the physiological responses downstream of TORC1 differentially regulated by TORC1 activity and if yes, how so, and 3) how does TOR mediate changes in survival and lifespan in response to various environmental manipulations, especially nutrients? In the rest of this article we will discuss the emerging evidence for this final question, which has become of growing interest and may also help illuminate the role of TOR in various human diseases.
The TOR signaling network mediates the protective effects of dietary restriction in different species
Aging is the single largest risk factor for disease in developed countries and arguably the single greatest challenge for biomedicine. Dietary restriction (DR) is the most robust environmental method to slow down aging in species as diverse as yeast, worms, fruit flies, rodents (Masoro, 2003) and recently even primates (Colman et al., 2009). DR also slows the progression of most age-related diseases, including some cancers, neurodegeneration and cardiovascular diseases (Masoro, 2003). DR is classically defined as a reduction of particular or total nutrient intake without causing malnutrition. In practice, DR is applied as an overall reduction of the caloric intake (calorie restriction), the restriction of a particular class of nutrients, temporal variations of food intake, or combinations thereof. Due to its robustness and strong conservation across species, identifying the molecular mechanisms that modulate lifespan upon DR holds great promise for providing insight into the mechanisms of aging. Examination of the molecular and genetic foundation of this protective response is moving at a feverish pace in the last few years. This has been made possible largely due to the short lifespan and powerful genetic tools that are available in S. cerevisiae, C. elegans and D. melanogaster. One of the proposed evolutionary mechanisms of DR is that under conditions of nutrient limitation, there is a shift in metabolic investment from reproduction and growth towards somatic maintenance to ensure extended survival (Holliday, 1989). Abundant evidence for TORC1 as a conserved nutrient sensor makes it an attractive candidate to mediate this switch between growth and somatic maintenance to extend lifespan by DR.
In S. cerevisiae, DR due to limitation of glucose has been shown to robustly extend lifespan. Replicative lifespan, measured by the number of replication events from a single mother yeast cell is increased when TOR activity is diminished. Furthermore, lowering glucose levels did not further extend lifespan of a tor1 deletion mutant (Kaeberlein et al., 2005). Hence, the TOR pathway mediates lifespan extension by DR imposed by limiting glucose concentrations in S. cerevisiae. Inhibition of TOR also extends yeast chronological lifespan (CLS), which measures the ability of stationary (in the absence of nutrients) cultures to maintain viability over time (Bonawitz et al., 2007; Powers et al., 2006).
Various components of the TOR signaling network have also been shown to mediate lifespan extension by DR imposed by different methods in C. elegans. Down-regulation of TOR expression by RNAi in C. elegans during adulthood results in lifespan extension (Vellai et al., 2003). This effect is independent of the FOXO ortholog DAF-16 (Vellai et al., 2003), a transcription factor that is required to mediate the lifespan extension caused by reduced insulin-like signaling (ILS) pathway activity (Guarente and Kenyon, 2000). Heterozygous daf-15 mutants, the worm ortholog of the mammalian protein raptor, display an extended lifespan, again lending credence to the hypothesis that partial inhibition of TORC1 activity suffices to extend life span in model organisms (Jia et al., 2004). Genes in the TOR pathway have been implicated in mediating the lifespan extension imposed by reducing bacterial food concentration in the diet of C. elegans (Chen et al., 2009b; Honjoh et al., 2009). In a model distinct from reducing available food, the eat-2 mutation, which causes pharyngeal pumping defects in C. elegans have been proposed to extend lifespan by eliciting a DR-like response (Lakowski and Hekimi, 1998). Additional inhibition of the TOR pathway and its downstream genes in an eat-2 mutant background does not further extend lifespan, suggesting that DR and lowered TORC1 signaling act by overlapping mechanisms (Chen et al., 2009b; Hansen et al., 2008).
In D. melanogaster, restriction of total food yields extended lifespan (Clancy et al., 2002; Mair et al., 2003; Rogina et al., 2002). A number of groups have shown that reducing yeast (the major source of protein in the fly diet) alone is also sufficient to extend lifespan in D. melanogaster (Chippindale et al., 1993; Kapahi et al., 2004; Mair et al., 2005). Overexpression of either Tsc1 or Tsc2, or dominant-negative forms of dTOR extend lifespan in D. melanogaster (Kapahi et al., 2004). Fat and muscle were observed to be the key tissues responsible for the lifespan extension effects of inhibiting TOR signaling in D. melanogaster (Kapahi et al., 2004). Under conditions of DR imposed by restricting dietary yeast, inhibition of the TOR pathway resulted in no further extension of lifespan. These findings support the idea that TOR mediates lifespan extension by DR in flies (Kapahi et al., 2004).
The role of the TOR signaling network in mediating mammalian aging phenotypes is also beginning to emerge. Long-lived Ames dwarf mice harbor mutations in a gene called Prop-1 that disrupts pituitary gland development (Bartke and Brown-Borg, 2004; Brown-Borg, 2009). It has been proposed that these mice show reduced protein synthesis possibly via inhibition of TOR signaling (Sharp and Bartke, 2005). These data support the notion that reduced signaling downstream of TOR extends lifespan in mice. The National Institute on Aging initiated the Interventions Testing Program (ITP) to evaluate drugs that putatively delay aging or prevent multiple forms of late-life disease in laboratory mice in three different laboratories. The administration of the TORC1 inhibitor rapamycin late in life (aged 600 days) was sufficient to cause a lifespan extension at all three sites (p<0.05) in both sexes, although there are sex-specific differences (males 9%, females 13%) (Harrison et al., 2009). A second study also demonstrated that rapamycin treatment, initiated past middle age (22–24 months old) increases lifespan in mice. This study also showed that under these conditions, rapamycin treatment can boost immune function and rejuvenate hematopoietic stem cells (Chen et al., 2009a). The role of TORC1 has also been tested in models of cellular senescence in culture. Notably, the effects of Doxorubicin (DOX), a DNA damaging agent mediating cellular senescence in normal human cells, were rescued by treatment with rapamycin (Demidenko and Blagosklonny, 2008). Together these data give promise to the notion that reduced TORC1 signaling may extend lifespan even in mammals.
These data emphasize the importance of TORC1 activity in mediating lifespan extension in multiple species and under different methods of nutrient manipulation. TORC2 plays a minor role in modulating lifespan. A study using C. elegans demonstrated that a mutant in Rictor shows lifespan shortening on feeding standard bacteria, but extends lifespan when fed certain types of nutrient-rich bacteria (Soukas et al., 2009). The discovery that the TOR signaling network mediates lifespan extension caused by nutrient modulation in invertebrate and vertebrate species provides a framework to examine the genetic and molecular basis of how DR extends lifespan. Strong inhibition of TORC1 early in life (embryogenesis or larval stages) drastically slows or even stops development. In contrast, inhibition of this pathway during adulthood (late life) extends lifespan. This is consistent with the predictions of the theory of antagonistic pleiotropy, which suggests that genes that are important for fitness early in life limit survival later in life (Williams, 1957). Various groups have now been able to mechanistically link various processes downstream of TORC1 to aging, which provides hope of achieving a more complete understanding of how growth, development and aging are linked with each other (Table 1, Figure 2).
Table 1.
Components of the TOR pathway that modulate lifespan in various species.
| Model system | Dietary manipulation/assay | Proteins implicated | Reference |
|---|---|---|---|
| M. musculus | None | TOR | (Harrison et al., 2009) |
| M. musculus | None | S6K | (Selman et al., 2009) |
| D. melanogaster | Yeast restriction | TOR, TSC1, TSC2, S6K | (Kapahi et al., 2004) |
| D. melanogaster | Yeast restriction | 4E-BP, Mitochondrial ETC | (Zid et al., 2009) |
| C. elegans | None | TOR | (Vellai et al., 2003) |
| C. elegans | None | Raptor | (Jia et al., 2004) |
| C. elegans | None | S6K, eIF4F complex | (Pan et al., 2007) |
| C. elegans | None | Translation factors, ribosomal proteins | (Hansen et al., 2007) |
| C. elegans | None | eIF4E | (Syntichaki et al., 2007) |
| C. elegans | None | Translation factors, ribosomal proteins | (Curran and Ruvkun, 2007) |
| C. elegans | Bacterial restriction | AMPK, DAF-16 | (Greer et al., 2007)) |
| C. elegans | None | Translation factors, ribosomal proteins | (Chen et al., 2007) |
| C. elegans | None | autophagy | (Hansen et al., 2008) |
| C. elegans | None | S6K, PHA-4 | (Sheaffer et al., 2008) |
| C. elegans | None | DAF-15, autophagy | (Toth et al., 2008) |
| C. elegans | Intermittent fasting | RHEB-1, DAF-16 | (Honjoh et al., 2009) |
| C. elegans | Bacterial restriction | S6K, HIF-1, EGL-9, IRE-1, Raptor | (Chen et al., 2009b) |
| C. elegans | None | TOR-interacting proteins | (Bell et al., 2009) |
| S. cerevisiae | Glucose restriction (replicative lifespan) | TOR, SCH9 | (Kaeberlein et al., 2005) |
| S. cerevisiae | Glucose restriction (chronological lifespan) | Mitochondrial ETC genes | (Bonawitz et al., 2007) |
| S. cerevisiae | Glucose restriction (replicative lifespan) | TOR, MSN2, MSN4, PNC1 | (Medvedik et al., 2007) |
| S. cerevisiae | Glucose restriction (replicative lifespan) | GCN4, 60S ribosomal subunit, TOR | (Steffen et al., 2008) |
| S. cerevisiae | Glucose restriction (chronological lifespan) | TOR, SCH9, RIM15, MSN2/4, GIS1 | (Wei et al., 2008) |
| S. cerevisiae | Glucose restriction (chronological lifespan) | TOR, SCH9, glycerol synthesis | (Wei et al., 2009) |
A list of studies that have demonstrated a role for genes in the TOR pathway or its interacting proteins to mediate lifespan extension by nutrient manipulation or under standard laboratory conditions in different model systems.
Figure 2. TOR pathway, a conserved mediator of lifespan extension in multiple species.

The diagram represents the genetic interactions in the TOR signaling network that execute lifespan extension in yeast, worms, flies and mice. The diagram summarizes the lifespan data for genes in the TOR signaling pathway or those that demonstrate a genetic interaction with mutants in the TOR pathway. The shaded area refers to components of the TOR pathway that show conserved effects on lifespan extension in different species. See main text for details.
Mediators of TORC1 that modulate lifespan
Protein synthesis
Inhibition of protein synthesis is one of the major effects mediated by reducing TOR pathway activity (Ma and Blenis, 2009; Shamji et al., 2003). The regulation of protein synthesis also plays a crucial role in keeping organismal growth and development in tune with environmental conditions (Sonenberg, 2000). Inhibition of protein synthesis by attenuating expression of various mRNA translation factors (Hansen et al., 2007; Henderson et al., 2006; Pan et al., 2007; Steffen et al., 2008; Syntichaki et al., 2007) or reducing protein in the diet (Chippindale et al., 1993; Kapahi et al., 2004; Miller et al., 2005; Orentreich et al., 1993; Richie et al., 1994) extends lifespan in multiple species. Furthermore, the rate of protein synthesis has been shown to inversely correlate with longevity across species (Finch, 1990; Munro, 1969), arguing for its role in determining species-specific lifespan.
The phenotypic overlap of DR and reduced translational initiation suggests that inhibition of protein synthesis is also likely to be important for the lifespan extension by DR. Given the high cost of mRNA translation in a cell, a shift of investment from continuing protein synthesis for growth towards more conservative somatic maintenance is conceivable under periods of DR. It was previously believed that total calorie intake was the most important aspect of extending lifespan by diet manipulation. However, a number of studies over the past two decades have demonstrated that restricting protein alone is sufficient for lifespan extension. In rodents, restricting the intake of single amino acids, such as methionine or tryptophan, yields an extended lifespan comparable to restricting ad libitum feeding (Miller et al., 2005; Orentreich et al., 1993; Richie et al., 1994; Zimmerman et al., 2003). Although total body weight was reduced in methionine-restricted animals, weight-normalized food intake was actually increased (Orentreich et al., 1993). These results suggest that a decrease in energy intake or expenditure on a body weight basis is not necessary for lifespan extension. In D. melanogaster, restriction of yeast, the major source of protein in the fly diet, is sufficient to extend lifespan (Chippindale et al., 1993; Kapahi et al., 2004; Mair et al., 2003). Reduction of protein fails to extend lifespan in rats (Masoro et al., 1989) and whether reduction of protein would extend lifespan in other species remains to be seen. Restriction of dietary protein may act directly on protein synthesis by reducing the pool of available amino acids, and/or by inhibiting growth-signaling pathways like TOR that respond to the presence of these macronutrients. The molecular mechanisms by which TORC1 activity modulates protein synthesis is a growing area of research (Ma and Blenis, 2009; Sonenberg and Hinnebusch, 2007; Wullschleger et al., 2006). Regulation of protein synthesis by TORC1 takes place at multiple levels including modulation of S6 kinase, mRNA translation initiation and ribosomal biogenesis. Modulation of mRNA translation is likely to be a major mechanism by which TORC1 modulates lifespan and is discussed below.
S6 kinase
Activation of ribosomal p70 S6 protein kinase (hereafter referred to as S6K) by phosphorylation is one of the most well-established downstream events in the TOR pathway. Loss of S6K results in slowed growth and reduced body size in multiple species including flies (Montagne et al., 1999), worms (Hansen et al., 2007; Pan et al., 2007), and mice (Um et al., 2004). Thus, similar to TORC1, inhibition of S6K also inhibits growth, but unlike TORC1, S6K is not essential for development. S6K modulates protein synthesis by a number of mechanisms which has been reviewed recently (Ma and Blenis, 2009). Inhibition of S6K has previously been shown to extend lifespan in D. melanogaster (Kapahi et al., 2004) and C. elegans (Hansen et al., 2007; Pan et al., 2007). In S. cerevisiae, mutations in SCH9, a proposed ortholog of S6K (Urban et al., 2007), have been known to significantly extend lifespan (Fabrizio et al., 2001; Kaeberlein et al., 2005). Recently, female but not male mice lacking S6K1 (the mouse genome encodes 2 paralogs, S6K1 and S6K2) have been shown to display increased lifespan and slowed progression of age-related pathologies (Selman et al., 2009). The sex-specific difference, although more pronounced, is reminiscent of the results from the rapamycin study and might point towards gender dependent effects of TORC1 signaling on longevity. Previously, it had also been shown that mice lacking S6K1 are resistant to diet-induced obesity and show improved insulin sensitivity (Um et al., 2004). These findings demonstrate that the longevity effects of inhibiting S6K are conserved in yeast, worms, flies and mice, supporting its role as a key player in the lifespan extension by reduced mammalian TORC1 signaling.
Regulation of mRNA translation initiation
mRNA translation proceeds through coordinated phases of initiation, elongation and termination, with initiation being the rate limiting step (Sonenberg, 2000). TORC1 enhances translation initiation of mRNA by inhibitory phosphorylation of the 4E-BP (eukaryotic initiation factor eIF4E binding protein) family of proteins (Shamji et al., 2003). Hypophosphorylated 4E-BPs act as translational repressors by binding to the translation initiation factor eIF4E. This inhibits the activity of the eIF4F translation initiation complex. eIF4E plays a critical role in initiation by recruiting mRNAs via their 5’UTRs. This is accomplished by regulating the association of the mRNA cap binding protein eIF4E to the scaffold protein eIF4G, both components of the eIF4F complex. eIF4G helps assemble the eIF4F complex by bridging the poly(A) binding proteins (PABPs) with eIF4E (Sonenberg, 2000). This leads to the circularization of mRNAs, which has a synergistic effect on enhancing the rate of translation (Sonenberg, 2000). TORC1 also modulates eIF4F complex activity by regulating the phosphorylation and degradation of eIF4G (Berset et al., 1998). Furthermore, given the role of TOR in activating S6K, which also modulates mRNA translation initiation by influencing the eIF3F complex (Ma and Blenis, 2009), TOR appears to influence mRNA translation initiation at multiple levels.
Recently, it has been shown that 4E-BP is an important mediator of the protective effects of DR in D. melanogaster. DR enforced by restriction of yeast in the diet requires intact 4E-BP to cause fully extended lifespan (Zid et al., 2009). DR also induces 4E-BP protein levels independently of FOXO, suggesting that nutrients regulate 4E-BP both by changes in gene expression and TORC1 dependent phosphorylation. Interestingly, under complete starvation conditions 4E-BP is also upregulated, though in a FOXO dependent manner, and is required for enhanced survival under these conditions (Teleman et al., 2005; Tettweiler et al., 2005). Overexpression of a gain of function form of 4E-BP, which has increased affinity for eIF4E (Miron et al., 2001), is sufficient to extend lifespan under rich nutrient conditions, but not under DR (Zid et al., 2009). Together these experiments justify the hypothesis that 4E-BP acts as a mediator of lifespan extension by DR and that a gain of function 4E-BP displays the hallmarks of a DR mimetic in D. melanogaster.
Studies from a number of groups have shown that inhibition of several components of the eIF4F complex and PABPs extends lifespan (Curran and Ruvkun, 2007; Hansen et al., 2007; Henderson et al., 2006; Long et al., 2002; Pan et al., 2007; Syntichaki et al., 2007). Inhibition of ifg-1, which encodes the C. elegans ortholog of eIF4G, during development causes larval arrest similar to that observed upon inhibition of TOR, while its reduced expression during adulthood increases lifespan and stress resistance (Hansen et al., 2007; Pan et al., 2007). Inhibition of eIF4E and eIF4G extends lifespan in C. elegans in a DAF-16-independent manner (Hansen et al., 2007; Pan et al., 2007; Syntichaki et al., 2007). These experiments suggest that reducing translational initiation is likely to slow aging by mechanisms distinct from those that extend lifespan upon inhibition of the insulin-like signaling pathway. Overexpression of eIF4E has also been shown to increase cellular senescence in mammalian cells as determined by β-galactosidase staining (Ruggero et al., 2004). These results support the notion that reduction of mRNA translation initiation by inhibition of the eIF4F complex might be a conserved mechanism to extend lifespan in multiple species.
Ribosomal biogenesis
Ribosomal proteins play a conserved and established role in protein synthesis and growth in various species. Ribosome production is an energetically expensive process and it has been proposed that TORC1 couples nutrient availability to ribosome production (Wullschleger et al., 2006). TORC1 signaling regulates ribosome biogenesis by multiple mechanisms which are not yet well understood (Guertin and Sabatini, 2007; Wullschleger et al., 2006). TORC1 is known to affect the transcription of mRNAs encoding ribosomal proteins by RNA polymerase II, as well as the transcription of ribosomal RNAs and transfer RNAs by RNA polymerase I and RNA polymerase III respectively (Wullschleger et al., 2006). In S. cerevisiae, it has been proposed that reduced ribosomal biogenesis downstream of the TOR signaling pathway mediates the effects of DR. This has been supported by the observation that loss of genes encoding the 60S ribosomal subunit extend lifespan (Steffen et al., 2008). Furthermore, the lifespan extension by DR, inhibition of TORC1 signaling, and inhibition of 60S ribosomal subunits employ overlapping mechanisms. Inhibition of various ribosomal subunits has also been shown to extend lifespan in C. elegans (Bell et al., 2009; Chen et al., 2007; Curran and Ruvkun, 2007; Hansen et al., 2007).
Mechanism of lifespan extension by modulating mRNA translation
How a reduction in protein synthesis extends lifespan is not entirely intuitive. This is especially hard to reconcile given the observation that there is a global decline in protein synthesis with age (Finch, 1990). Two models may potentially explain the beneficial effects of inhibiting protein synthesis. One possibility is that a global decrease in protein synthesis may improve the fidelity of protein synthesis. Furthermore, a decrease of overall mRNA translation could also decrease the overall burden of misfolded proteins by reducing the protein traffic intracellularly. Another possibility is that the mRNA translation of specific mRNAs may be preserved or even enhanced under conditions of global inhibition of protein synthesis, and that this may slow aging. These two models are not mutually exclusive and accumulating evidence favoring the latter model is discussed below.
Previous studies have pointed out the importance of regulating mRNA translation, especially under environmental stresses (Sonenberg, 2000; Sonenberg and Hinnebusch, 2007). One well-studied example is that of the transcription factor GCN4. Upon starvation, while the global rate of protein synthesis decreases, production of GCN4 protein increases by an enhancement in the ribosomal loading for GCN4 mRNA. Mechanistically, this is driven by short upstream open reading frames in the 5’UTR of this gene (Hinnebusch, 2005; Tzamarias et al., 1989). GCN4 has been shown to contribute to the increased lifespan caused by RNAi against 60S ribosomal subunits (Steffen et al., 2008).
Methods to assess genome wide changes in translation state are likely to provide useful insights into how changes in mRNA translation modulate lifespan. Genome wide translational profiling examines the number of polysomes bound to each individual mRNA. Given that mRNA translation initiation is the major rate-limiting step in mRNA translation, this indicates the efficiency with which each mRNA is translated (Serikawa et al., 2003; Zong et al., 1999). Genome wide translational changes revealed an increase in the ribosomal binding of certain mRNAs, though there was an overall decrease of mRNA translation under DR in D. melanogaster (Zid et al., 2009). Surprisingly, a number of nuclear-encoded mitochondrial genes, including those encoding the electron transport chain (ETC) complexes I and IV and mitochondrial ribosomal proteins, displayed increased ribosomal binding and mRNA translation (Zid et al., 2009). The enhanced ribosomal loading of nuclear-encoded mitochondrial genes was dependent on their simple 5’UTR structures. These results suggest a novel mode of regulation of nuclear-encoded mitochondrial genes by enhanced mRNA translation under DR. However, the precise mechanisms by which the translation machinery upregulates these genes remains to be elucidated.
The effects of protein synthesis inhibition in different model systems provide a novel paradigm to explore the mechanisms of aging and DR. However, in addition to assessing the mRNA translation state of each mRNA, information on the half lives of each mRNA, their elongation rates and their turnover once converted to protein, will provide novel insights into how modulation of mRNA translation and protein synthesis alter the rate of aging.
Stress responses and transcription factors
DR and a number of long-lived mutants enhance resistance to various environmental stresses (Martin et al., 1996; Masoro, 2003). The modulation of stress pathways has been well established to contribute to lifespan extension in multiple species (Martin et al., 1996). As mentioned above, a similar array of stresses also modulates TORC1 activity. This poses the question whether reduced TORC1 activity triggers a stress resistance response. Inhibition of the TOR signaling network and also a number of translation factor genes enhances resistance to various environmental stresses (Hansen et al., 2007; Kaeberlein et al., 2005; Pan et al., 2007; Powers et al., 2006). Evidence from both C. elegans and S. cerevisae suggest that transcription factors involved in mediating stress responses are regulated by TORC1 signaling and mediate its effects on lifespan extension.
In C. elegans, the transcription factor PHA-4 plays an essential role in the embryonic development of the foregut (Mango, 2009). The mammalian orthologs of PHA-4, the Foxa transcription factors Foxa1, Foxa2 and Foxa3, also play important roles during development, and act later in life to regulate glucagon production and glucose homeostasis in response to fasting (Mair and Dillin, 2008). The interaction between PHA-4 and TOR signaling have been uncovered by the finding that inactivation of let-363/tor or rsks-1/s6k can suppress the lethality associated with pha-4 mutants in C. elegans (Sheaffer et al., 2008) Furthermore, lifespan extension by inhibition of S6K was dependent on pha-4 (Sheaffer et al., 2008). PHA-4 has previously been described as a critical regulator of lifespan extension mediated by DR but not other longevity pathways in C. elegans (Panowski et al., 2007). Together, these findings argue for the role of S6K in mediating lifespan extension by DR through PHA-4 dependent mechanisms.
AMPK is the central player that becomes activated upon an increased AMP/ATP ratio, indicating a reduced energy load in the cell. While biochemical evidence places AMPK upstream to TORC1 (Figure 1), their relationship in lifespan regulation is more complex (Figure 2). It has been shown that AMPK acts downstream of S6K and mediates the S6K-dependent effects on body size and lifespan (Selman et al., 2009). In C. elegans, loss of AMPK in a S6K mutant background revert the body size and fecundity defects of the S6K mutant. Though biochemical data demonstrating that AMPK is a direct target of S6K is lacking, the above results place AMPK downstream of S6K in terms of mediating its effects on growth as well as lifespan. However, this apparently counterintuitive result can be reconciled by findings that both in mice and worms, loss of S6K causes increased AMPK activity (Aguilar et al., 2007; Selman et al., 2009). Hence, when DR causes a reduction of TORC1 and S6K, AMPK may become active and lead to sustained TORC1 inhibition. This model suggests that AMPK, TORC1 and S6K1 constitute a feedback signaling loop that resets its activity upon DR to re-direct growth, metabolism and lifespan.
AMPK has also been proposed to be an important mediator of the lifespan extension by DR imposed by dilution of bacteria in the food (Greer et al., 2007). Furthermore, the AMPK agonist, metformin, extends lifespan in at least one genetic strain of mice (Anisimov et al., 2008). Despite the tight biochemical and phenotypic linkage of AMPK and TORC1, it is likely that AMPK also receives inputs independently of TORC1/S6K to influence lifespan. In support of this idea, AMPK has been proposed to partially mediate the lifespan extension observed in the long-lived C. elegans daf-2 mutant (Apfeld et al., 2004). Furthermore, links between AMPK and the ILS pathway have been demonstrated by the finding that human and worm AMPK directly phosphorylates the FOXO transcription factor. DAF-16, the worm ortholog of FOXO is required for the lifespan extension by activation of AMPK (Greer et al., 2007) and also the maximal lifespan extension by DR in some, but not all DR regimes in C. elegans (Greer and Brunet, 2009).
The regulation of the stress-responsive transcription factor HIF-1α is under the control of TORC1 activity at both transcriptional and translational levels (Bernardi et al., 2006; Hui et al., 2006). This idea is supported by findings in cell culture studies showing that rapamycin treated cells fail to adapt to hypoxia (Thomas et al., 2006). Low oxygen concentration results in the stabilization of the transcription factor HIF-1α and the activation of the hypoxic response (Kaelin and Ratcliffe, 2008). HIF-1α helps cells adapt to low oxygen stress by regulating angiogenesis, glycolysis, and cell survival (Semenza, 2000). A recent study suggests that in addition to being responsive to oxygen levels, HIF-1 is also responsive to nutrients and mediates the effects of DR on lifespan extension in C. elegans. Genetic epistasis analysis places HIF-1 downstream of S6K to mediate lifespan extension. A mutation in hif-1 extends lifespan on rich nutrient conditions but does not cause further lifespan extension under DR, whereas the egl-9 mutant, with elevated HIF-1, fails to show maximal lifespan extension under DR. Thus, there is an inverse correlation between HIF-1 activities and lifespan, suggesting that maximal lifespan extension by DR is mediated by HIF-1 in C. elegans (Chen et al., 2009b). This study also found that HIF-1 activity in muscle and specific neurons was critical for mediating its effects on DR (Chen et al., 2009b). Interestingly, an increase in HIF-1 levels in C. elegans has also been implicated in lifespan extension, although by mechanisms distinct from DR (Mehta et al., 2009; Zhang et al., 2009). The reasons for this remain unclear (Kaeberlein and Kapahi, 2009) but may involve the action of HIF-1 in different tissues for its differential effects on lifespan. These findings open a new area of investigation to examine the link between nutrients, oxygen tension, cancer and aging.
In yeast, TORC1 inhibition leads to enhanced resistance to stress and nuclear translocation of Msn2, a stress-induced transcription factor (Powers et al., 2006). Msn2/4 were also found to be translocated to the nucleus and to contribute to lifespan extension upon inhibition of TOR by enhancing the levels of the nicotinamidase gene, PNC1 (Medvedik et al., 2007). The serine/threonine kinase Rim15 positively regulates the stress response transcription factors Gis1 and Msn2/4 and is required for yeast chronological lifespan extension caused by deficiencies in Tor1, Sch9, and by DR. A recent study in S. cerevisiae also found that DR and inhibition of the TOR pathway enhances stress resistance by switching metabolism to enhance glycerol synthesis (Wei et al., 2009). Deletion of the glycerol biosynthesis genes that were up-regulated in long-lived TOR pathway mutants was sufficient to reverse the chronological lifespan extension and enhanced stress resistance, suggesting that glycerol production mediates the enhanced stress resistance and increased chronological lifespan in yeast (Wei et al., 2009). Together, these experiments support the idea that inhibition of TORC1 plays an important role in mediating the switch of cellular resources from growth and reproduction towards somatic maintenance and lifespan extension.
Autophagy
Autophagy is defined as a highly regulated cellular starvation response to maintain essential nutrient levels and viability. During macroautophagy, various cytoplasmic components, including organelles, are enclosed within a double-membrane structure (the autophagosome) and delivered to the vacuole (in S. cerevisiae) or lysosome (in other eukaryotic cells) for degradation and recycling of the degradation products (Cecconi and Levine, 2008; Diaz-Troya et al., 2008). The role of autophagy in modulating lifespan was first identified when the extended lifespan of the C. elegans mutant in daf-2, which encodes an insulin like receptor, was significantly abrogated by inhibition of the autophagy gene bec-1 (Melendez et al., 2003). As inhibition of autophagy does not compromise the lifespan of control animals in both worms and flies (Hansen et al., 2008; Melendez et al., 2003; Ren et al., 2009), this observation supports the idea that enhanced autophagy may extend lifespan. Inhibition of TORC1 enhances autophagy and provides an attractive mechanism to explain the effects of reduced TORC1 activity on lifespan. In fact, autophagy is also required for the lifespan extension mediated by DR or inhibition of TORC1 (Hansen et al., 2008; Toth et al., 2008). Additionally, lifespan extension by the eat-2 mutation can be suppressed by inhibition of autophagy genes using RNAi (Hansen et al., 2008; Jia and Levine, 2007). Increased autophagy has been observed under DR and requires the FOXA transcription factor PHA-4 (Hansen et al., 2008), which is also a critical regulator of lifespan extension mediated by DR (Panowski et al., 2007) and in S6K mutants (Sheaffer et al., 2008). Together these experiments implicate an important role for autophagy in lifespan extension by DR. Future experiments examining the role of autophagy in specific tissues will be especially useful to our understanding its effects on lifespan.
Endoplasmic Reticulum (ER) stress
ER stress is caused by a mismatch between the load of unfolded/misfolded proteins within the ER and the capacity of the cellular machinery to cope with it (Ron and Walter, 2007). An increase in ER stress and activation of the unfolded protein response (UPR) in mammalian cells has been observed in response to excess nutrients (Ozcan et al., 2004) or enhanced TORC1 signaling due to loss of TSC1 or TSC2 (Ozcan et al., 2008). Genes that help cope with ER stress have been shown to mediate lifespan extension in both S. cerevisiae (Steffen et al., 2008) and C. elegans (Chen et al., 2009b). One possibility is that GCN4, which mediates the lifespan extension effects of DR, enhances survival by modulating ER signaling (Steffen et al., 2008). This is supported by the findings that GCN4 has been shown to regulate a number of genes required for UPR in S. cerevisiae (Patil et al., 2004). Consistent with a role for the ER signaling pathway in C. elegans aging, ire-1 was found to be required for lifespan determination by DR and by loss of hif-1 function (Chen et al., 2009b). IRE-1 is an ER transmembrane protein with kinase and RNAse activities, which senses misfolded proteins in the ER lumen and responds by splicing the xbp-1 mRNA. This allows the translation of functional XBP-1 protein which regulates target genes required for increased ER stress resistance (Ron and Walter, 2007). These studies open a rich area of investigation to examine the mechanisms by which ER stress signaling, which has been linked with obesity and diabetes, mediates DR dependent lifespan extension.
Metabolism
DR has significant effects on metabolism, which has been suggested to modulate the rate of aging (Masoro, 2003). Two studies have shown that increased mitochondrial function by either inhibition of TORC1 or by DR contributes to the lifespan extension in S. cerevisiae and D. melanogaster. This is a surprising finding given that it is widely believed that enhanced mitochondrial function would result in increased oxidative damage by ROS (reactive oxygen species). The first study proposes that deletion of the TOR1 gene extends chronological lifespan in S. cerevisiae by increasing mitochondrial respiration via enhanced translation of mtDNA-encoded oxidative phosphorylation complexes (Bonawitz et al., 2007). The increased chronological lifespan upon deletion of TOR1 was reversed by inhibition of ETC function. The second study showed that in D. melanogaster, DR causes increased translation of nuclear-encoded mitochondrial genes, encoding for ETC and ribosomal proteins, thereby enhancing mitochondrial density and function (Zid et al., 2009). The role of mitochondrial function in mediating lifespan extension in D. melanogaster was confirmed by the observation that inhibition of complex I throughout the whole body abrogated the lifespan extension by DR (Zid et al., 2009).
These observations suggest that the translational regulation of mitochondrial function and its protective effects during DR may be conserved in different species. Surprisingly, and in contrast to these findings, it has been observed that in C. elegans, inhibition of components of the ETC also lead to lifespan extension (Aguilaniu et al., 2005). These observations are counterintuitive to the proposed idea that a deficiency of ETC function with age, observed in multiple species, may be responsible for organismal aging (McCarroll et al., 2004). One possible explanation reconciling these contradictory findings is that lifespan extension by reduction of ETC function is tissue specific. In support of this idea it has recently been shown that inhibition of ETC in neurons can extend lifespan even in D. melanogaster (Copeland et al., 2009). Hence, it is conceivable that age-related decline in mitochondrial function may contribute to aging by action in non-neuronal tissues.
An outstanding question is how increased reliance on mitochondrial respiration extends lifespan. The activity level of TORC1 might regulate the switch between glycolysis and mitochondrial respiration for the generation of ATP. Highly active TORC1 promotes high levels of HIF-1, shifting the cell towards glycolysis. In contrast, TORC1 inhibition favors mitochondrial ATP generation. The subsequent increase in mitochondrial oxygen consumption might actually limit intracellular oxygen and ROS-mediated damage associated with aging (Bonawitz et al., 2007). Alternatively, the enhanced translation of mitochondrial ETC genes serves as a protective mechanism not only by increasing mitochondrial efficiency but also by maintaining the function of the ETC and, hence, ATP production, which is known to decline with age (McCarroll et al., 2004). The shift of cancer cells away from mitochondrial respiration towards glycolysis for ATP generation even in the presence of oxygen has been termed the Warburg effect (Warburg, 1956). A causal role for mitochondrial respiration in tumorigenesis is supported by findings that succinate dehydrogenase and fumarate hydratase are tumor suppressor genes (King et al., 2006). However, future work in this area is required to identify the mechanisms by which the mitochondrial function exerts protective effects to slow aging and cancer progression.
Summary and future outlook
We have summarized the ample evidence for the role of nutrient and stress sensing by the TOR signaling network in mediating responses to DR. Strong evidence supports a regulatory role for reduced TOR signaling network activity to extend lifespan, as this is observed in all model organisms tested so far (Figure 2). This provides a strong framework and excellent tools for exploring the link between nutrient limitation and lifespan extension. A conserved role for many components of this network including TOR, S6K and AMPK is emerging. This body of work provides an array of excellent putative drug targets for extending lifespan and slowing age-related diseases.
However, future work in this area examining the role of different types of nutrients in various species to modulate lifespan is needed to better assess the role of TOR in lifespan extension by dietary restriction. Furthermore, it remains to be seen which downstream effectors of TOR are key drivers of the longevity phenotype and which ones elicit only minor effects. The role of various tissues and their communication to determine organismal lifespan also remain poorly understood. Moreover, a number of pathways that modulate aging are being discovered, which display effects on lifespan seemingly independent of TORC1. The central challenge of aging research remains to flesh out a unified picture of mechanisms determining aging. Further analyses of TOR and other conserved pathways will shed light on the link between diet and various age-related diseases such as cancer, neurodegeneration and diabetes in humans.
Acknowledgments
We regret not being able to cite all of the relevant references owing to space limitations. This work was funded by grants from the Ellison Medical Foundation, American Foundation for aging research, Hillblom foundation, a Nathan Shock Startup award and the NIH/NIA (PK), and REAC Award (LK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilaniu H, Durieux J, Dillin A. Metabolism, ubiquinone synthesis, and longevity. Genes Dev. 2005;19:2399–2406. doi: 10.1101/gad.1366505. [DOI] [PubMed] [Google Scholar]
- Aguilar V, Alliouachene S, Sotiropoulos A, Sobering A, Athea Y, Djouadi F, Miraux S, Thiaudiere E, Foretz M, Viollet B, Diolez P, Bastin J, Benit P, Rustin P, Carling D, Sandri M, Ventura-Clapier R, Pende M. S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab. 2007;5:476–487. doi: 10.1016/j.cmet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Pearce LR, Garcia-Martinez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal. 2009;2:pe27. doi: 10.1126/scisignal.267pe27. [DOI] [PubMed] [Google Scholar]
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, Semenchenko AV. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–2773. doi: 10.4161/cc.7.17.6625. [DOI] [PubMed] [Google Scholar]
- Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A, Brown-Borg H. Life extension in the dwarf mouse. Curr Top Dev Biol. 2004;63:189–225. doi: 10.1016/S0070-2153(04)63006-7. [DOI] [PubMed] [Google Scholar]
- Bell R, Hubbard A, Chettier R, Chen D, Miller JP, Kapahi P, Tarnopolsky M, Sahasrabuhde S, Melov S, Hughes RE. A human protein interaction network shows conservation of aging processes between human and invertebrate species. PLoS Genet. 2009;5:e1000414. doi: 10.1371/journal.pgen.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Ruegg MA. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;8:411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Berset C, Trachsel H, Altmann M. The TOR (target of rapamycin) signal transduction pathway regulates the stability of translation initiation factor eIF4G in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1998;95:4264–4269. doi: 10.1073/pnas.95.8.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5:265–277. doi: 10.1016/j.cmet.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM. Hormonal control of aging in rodents: the somatotropic axis. Mol Cell Endocrinol. 2009;299:64–71. doi: 10.1016/j.mce.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15:344–357. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009a;2:ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Pan KZ, Palter JE, Kapahi P. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell. 2007;6:525–533. doi: 10.1111/j.1474-9726.2007.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009b;5:e1000486. doi: 10.1371/journal.pgen.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chippindale AK, Leroi AM, Kim SB, Rose MR. Phenotypic plasticity and selection in Drosophila life-history evolution. I. Nutrition and the cost of reproduction. J Evol Biology. 1993;6:171–193. [Google Scholar]
- Clancy DJ, Gems D, Hafen E, Leevers SJ, Partridge L. Dietary restriction in long-lived dwarf flies. Science. 2002;296:319. doi: 10.1126/science.1069366. [DOI] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–3361. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2002;99:4319–4324. doi: 10.1073/pnas.261702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–865. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- Finch CE. Longevity, Senescence, and the Genome. Chicago: University of Chicago Press; 1990. [Google Scholar]
- Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009;8:113–127. doi: 10.1111/j.1474-9726.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–361. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Bonafe M, Johnson TE. daf-16 protects the nematode Caenorhabditis elegans during food deprivation. J Gerontol A Biol Sci Med Sci. 2006;61:444–460. doi: 10.1093/gerona/61.5.444. [DOI] [PubMed] [Google Scholar]
- Hentges KE, Sirry B, Gingeras AC, Sarbassov D, Sonenberg N, Sabatini D, Peterson AS. FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc Natl Acad Sci U S A. 2001;98:13796–13801. doi: 10.1073/pnas.241184198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Holliday R. Food, reproduction and longevity: is the extended lifespan of calorie-restricted animals an evolutionary adaptation? Bioessays. 1989;10:125–127. doi: 10.1002/bies.950100408. [DOI] [PubMed] [Google Scholar]
- Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2009;457:726–730. doi: 10.1038/nature07583. [DOI] [PubMed] [Google Scholar]
- Hui AS, Bauer AL, Striet JB, Schnell PO, Czyzyk-Krzeska MF. Calcium signaling stimulates translation of HIF-alpha during hypoxia. Faseb J. 2006;20:466–475. doi: 10.1096/fj.05-5086com. [DOI] [PubMed] [Google Scholar]
- Inoki K, Guan KL. Complexity of the TOR signaling network. Trends Cell Biol. 2006;16:206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Kapahi P. The hypoxic response and aging. Cell Cycle. 2009;8:2324. doi: 10.4161/cc.8.15.9126. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- Kuruvilla FG, Shamji AF, Schreiber SL. Carbon- and nitrogen-quality signaling to translation are mediated by distinct GATA-type transcription factors. Proc Natl Acad Sci U S A. 2001;98:7283–7288. doi: 10.1073/pnas.121186898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–451. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Long X, Spycher C, Han ZS, Rose AM, Muller F, Avruch J. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol. 2002;12:1448–1461. doi: 10.1016/s0960-9822(02)01091-6. [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- Mair W, Goymer P, Pletcher SD, Partridge L. Demography of dietary restriction and death in Drosophila. Science. 2003;301:1731–1733. doi: 10.1126/science.1086016. [DOI] [PubMed] [Google Scholar]
- Mair W, Piper MD, Partridge L. Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biol. 2005;3:e223. doi: 10.1371/journal.pbio.0030223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mango SE. The molecular basis of organ formation: insights from the C. elegans foregut. Annu Rev Cell Dev Biol. 2009;25:597–628. doi: 10.1146/annurev.cellbio.24.110707.175411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DE, Hall MN. The expanding TOR signaling network. Curr Opin Cell Biol. 2005;17:158–166. doi: 10.1016/j.ceb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Martin GM, Austad SN, Johnson TE. Genetic analysis of ageing: role of oxidative damage and environmental stresses. Nat Genet. 1996;13:25–34. doi: 10.1038/ng0596-25. [DOI] [PubMed] [Google Scholar]
- Masoro EJ. Subfield history: caloric restriction, slowing aging, and extending life. Sci Aging Knowledge Environ. 2003;2003:RE2. doi: 10.1126/sageke.2003.8.re2. [DOI] [PubMed] [Google Scholar]
- Masoro EJ, Iwasaki K, Gleiser CA, McMahan CA, Seo EJ, Yu BP. Dietary modulation of the progression of nephropathy in aging rats: an evaluation of the importance of protein. Am J Clin Nutr. 1989;49:1217–1227. doi: 10.1093/ajcn/49.6.1217. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004;36:197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e261. doi: 10.1371/journal.pbio.0050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal Regulation of the Hypoxic Response Modulates Aging in C. elegans. Science. 2009 doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell. 2005;4:119–125. doi: 10.1111/j.1474-9726.2005.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron M, Verdu J, Lachance PE, Birnbaum MJ, Lasko PF, Sonenberg N. The translational inhibitor 4E-BP is an effector of PI(3)K/Akt signalling and cell growth in Drosophila. Nat Cell Biol. 2001;3:596–601. doi: 10.1038/35078571. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Munro HN. Evolution of protein metabolism in mammals. In: Munro HN, Allison JB, editors. Mammalian Protein Metabolism. Vol. 3. 1969. pp. 133–182. [Google Scholar]
- Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710–6718. doi: 10.1128/MCB.24.15.6710-6718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 2000;14:2689–2694. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low methionine ingestion by rats extends life span. J Nutr. 1993;123:269–274. doi: 10.1093/jn/123.2.269. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, Hotamisligil GS. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007;6:111–119. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- Patil CK, Li H, Walter P. Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2004;2:E246. doi: 10.1371/journal.pbio.0020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–2892. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Finkel SE, Tower J. Conditional inhibition of autophagy genes in adult Drosophila impairs immunity without compromising longevity. Exp Gerontol. 2009;44:228–235. doi: 10.1016/j.exger.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richie JP, Jr, Leutzinger Y, Parthasarathy S, Malloy V, Orentreich N, Zimmerman JA. Methionine restriction increases blood glutathione and longevity in F344 rats. Faseb J. 1994;8:1302–1307. doi: 10.1096/fasebj.8.15.8001743. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL, Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 2002;298:1745. doi: 10.1126/science.1078986. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The Translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nature. 2004;10:484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev. 2000;14:1983–1991. [PubMed] [Google Scholar]
- Serikawa KA, Xu XL, MacKay VL, Law GL, Zong Q, Zhao LP, Bumgarner R, Morris DR. The Transcriptome and Its Translation during Recovery from Cell Cycle Arrest in Saccharomyces cerevisiae. Mol Cell Proteomics. 2003;2:191–204. doi: 10.1074/mcp.D200002-MCP200. [DOI] [PubMed] [Google Scholar]
- Shamji AF, Kuruvilla FG, Schreiber SL. Partitioning the transcriptional program induced by rapamycin among the effectors of the Tor proteins. Curr Biol. 2000;10:1574–1581. doi: 10.1016/s0960-9822(00)00866-6. [DOI] [PubMed] [Google Scholar]
- Shamji AF, Nghiem P, Schreiber SL. Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell. 2003;12:271–280. doi: 10.1016/j.molcel.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Sharp ZD, Bartke A. Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J Gerontol A Biol Sci Med Sci. 2005;60:293–300. doi: 10.1093/gerona/60.3.293. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Sheaffer KL, Updike DL, Mango SE. The Target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr Biol. 2008;18:1355–1364. doi: 10.1016/j.cub.2008.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hershey JWB, Mathews BM. Translational control of gene expression. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell. 2007;28:721–729. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 2009;23:496–511. doi: 10.1101/gad.1775409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BN, Pak DN, Fields S, Kennedy BK, Kaeberlein M. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell. 2008;133:292–302. doi: 10.1016/j.cell.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007;445:922–926. doi: 10.1038/nature05603. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Chen YW, Cohen SM. 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev. 2005;19:1844–1848. doi: 10.1101/gad.341505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettweiler G, Miron M, Jenkins M, Sonenberg N, Lasko PF. Starvation and oxidative stress resistance in Drosophila are mediated through the eIF4E-binding protein, d4E-BP. Genes Dev. 2005;19:1840–1843. doi: 10.1101/gad.1311805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K, Orosz L, Kovacs AL, Csikos G, Sass M, Vellai T. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008;4:330–338. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- Tzamarias D, Roussou I, Thireos G. Coupling of GCN4 mRNA translational activation with decreased rates of polypeptide chain initiation. Cell. 1989;57:947–954. doi: 10.1016/0092-8674(89)90333-4. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008;4:e13. doi: 10.1371/journal.pgen.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Madia F, Hu J, Ge H, Li LM, Longo VD. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet. 2009;5:e1000467. doi: 10.1371/journal.pgen.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution. 1957;11:398–411. [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14:2712–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009;4:e6348. doi: 10.1371/journal.pone.0006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JA, Malloy V, Krajcik R, Orentreich N. Nutritional control of aging. Exp Gerontol. 2003;38:47–52. doi: 10.1016/s0531-5565(02)00149-3. [DOI] [PubMed] [Google Scholar]
- Zong Q, Schummer M, Hood L, Morris DR. Messenger RNA translation state: the second dimension of high-throughput expression screening. Proc Natl Acad Sci U S A. 1999;96:10632–10636. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]