
Abstract
The role of androgen receptor (AR) activation and expression is well understood in prostate cancer. In breast cancer, expression and activation of AR is increasingly recognized for its role in cancer development and its importance in promoting cell growth in the presence or absence of estrogen. As both prostate and breast cancers often share a reliance on nuclear hormone signaling, there is increasing appreciation of the overlap between activated cellular pathways in these cancers in response to androgen signaling. Targeting of the androgen receptor as a monotherapy or in combination with other conventional therapies has proven to be an effective clinical strategy for the treatment of patients with prostate cancer, and these therapeutic strategies are increasingly being investigated in breast cancer. This overlap suggests that targeting androgens and AR signaling in other cancer types may also be effective. This manuscript will review the role of AR in various cellular processes that promote tumorigenesis and metastasis, first in prostate cancer and then in breast cancer, as well as discuss ongoing efforts to target AR for the more effective treatment and prevention of cancer, especially breast cancer.
Similar content being viewed by others


Introduction
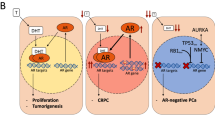
Androgens are expressed at different levels in men and women, and while they are important for proper development, they can also drive tumor growth. Most notably, the role of the androgen receptor (AR) in prostate cancer has been extensively studied. Recent data suggest that AR signaling may also be important in breast cancer, glioblastoma, and additional tumor types with AR expression1. In order to develop effective treatment strategies for patients with each of these cancer types, it is important to understand how AR is functioning similarly and differently to drive tumor growth.
AR belongs to the Type I class of nuclear hormone transcription factors along with the estrogen receptor (ER), progesterone receptor (PR), and glucocorticoid receptor (GR)2. As is characteristic of this type of receptor, inactive forms of AR are located in the cytoplasm, bound to heat shock proteins (HSPs)3. The HSPs are responsible for proper protein folding, prevention of misfolding and maintaining 3D protein structure during events of cellular stress4,5. AR, like other receptors in this family, is activated by the binding of androgen molecules to its ligand binding domain (LBD). Androgen binding results in AR homodimerization and translocation into the nucleus, where AR binds to androgen response elements (AREs) resulting in activation and transcription of a variety of downstream genes. Binding of AR results in the activation of diverse signaling pathways, including multiple signaling pathways that have been implicated in cancer, including the PI3K/AKT pathway6. AR also contributes to cell growth and proliferation differently in the context or absence of ER, and AR has an influence on cell cycle and DNA damage repair. Further, AR has non-genomic functions that can influence cell growth, migration, metastasis, and apoptosis. Due to its many downstream effects, antiandrogen therapies have long been of therapeutic interest along with combining AR antagonism with conventional chemotherapy or radiation therapy.
Gene expression and hormone receptor function
The AR has been well characterized as a key driver for the growth of prostate cancer in men. In this context, castration or androgen deprivation therapy (ADT) is a first line of therapy for men with metastatic prostate cancer. Despite the efficacy of ADT, resistance is near-universal. In some men, resistance can be mediated by AR amplification7, and others develop mutations in the LBD of AR in response to antiandrogen treatment8. These mutations can render cells refractory to androgen deprivation as there is constitutive AR activation, even in the absence of androgens. This results in activation of AR including AR binding to AREs and constitutive AR-regulated gene expression. More recently, a role for AR in the progression of breast cancer has been described. While AR’s function has not been fully characterized in breast cancer, work done in prostate cancer informs the potential function of AR in breast cancer.
Similar to the role that AR plays in prostate cancer development and progression, the ER has been recognized for the integral role that it plays in driving the development of the majority of breast cancers9. Breast cancers that express the ER (ER+) grow in response to the presence of estrogen and are more responsive to endocrine ablation10,11. This understanding led to some of the first “molecularly targeted therapies” that established the use of aromatase inhibitors (AIs) or selective estrogen receptor modulators (SERMs), which block the production and signaling of estrogen12. AIs and SERMs have been used as effective therapies for women with tumors that express ER13,14. Despite having identified the presence of AR expression in breast cancer many years ago15, little is known about the role of androgen signaling in breast cancer, though its importance as a potentially effective therapeutic target is increasingly appreciated and will be discussed herein. We begin with a review of the various processes known to be mediated by AR signaling, as recent studies have shed light on the role of AR with other pathways known to be abrogated in cancer.
Transcription factor and protein interactions
AR and FOXA1
Prostate cancer
FOXA1 is a transcription factor which plays an important role in aiding the binding of hormone receptors to their target DNA16. More recently, three distinct classes of alterations in FOXA1 have been described in prostate cancer, each with unique structural and phenotypic consequences17. The Class-1 activating mutations originate in early prostate cancer without alterations in ETS or SPOP and are found in the wing-2 region of the DNA-binding forkhead domain. Functionally these mutations allow for enhanced chromatin mobility and binding frequency and strongly transactivate a luminal AR program. The second class of activating mutations are found in metastatic prostate cancer and are characterized by a truncated C-terminal domain. These mutations increase FOXA1 DNA affinity and promote metastasis by activating the Wnt pathway through TLE3 inactivation. The final class of FOXA1 genomic rearrangements are characterized by duplications and translocations within the FOXA1 locus that reconfigure regulatory elements (FOXA1 mastermind elements) to drive overexpression of FOXA1. This third class of alterations is found primarily in metastatic prostate cancer and further underscores the interaction and significance of AR and FOXA1 protein interactions17. Similar classes of alterations also been observed in breast cancer1. In prostate cancer, FOXA1 also influences the ability of AR to bind DNA and control cell cycle progression. FOXA1 binds to genes necessary for growth of castration-resistant prostate cancer (CRPC), suggesting that FOXA1 is responsible for driving cell cycle progression in CRPC both from G1 to S and G2 to M18. FOXA1 also facilitates cell cycle progression from G2 to M by acting as a cofactor for AR18. Unsurprisingly, there is also significant overlap between genomic binding sites occupied by AR and FOXA119. While AR binds to many DNA regions independent of FOXA1, DNA-binding sites often require the presence of FOXA1 for AR recruitment19. Therefore, loss of FOXA1 results in the inability of AR to bind many DNA loci19. Using H3K4me2 ChIP analyses, Sahu et al. found that there were H3K4me2 marks at ~70% of sites shared by AR and FOXA119. Furthermore, staining of FOXA1 has been shown to correlate with disease outcomes in prostate cancer patients, where even with high AR staining, low FOXA1 is associated with good prognoses, and strong FOXA1 staining correlates with poor prognoses19, indicating that FOXA1 may have an important effect on AR signaling and tumor progression. Levels of FOXA1 are also elevated in prostate tumors and metastases, and overexpression of FOXA1 in prostate cancer cell lines results in increased AR binding at novel sites that have high chromatin accessibility20. These results suggest that increased levels of FOXA1 enhance AR binding to novel sites in order to facilitate cancer cell growth20 and implicate the importance of FOXA1 on AR function and tumor progression.
Breast cancer
FOXA1 is also essential for the growth of ER+ breast cancer cell lines21. Similar to prostate cancer, ChIP-seq studies have shown that there is extensive overlap between locations of AR and FOXA1 binding in breast cancer cells22. The function of AR in breast cancer is also dependent upon FOXA1, as silencing of FOXA1 inhibits AR binding of target DNA as well as cell growth22. In addition, FOXA1 functions as a transcription factor, playing an important role in aiding binding of hormone receptors, including ER and AR, to their target DNA16,23. When expressed with AR, FOXA1 may direct AR binding at sites of ER binding in luminal tumors24. Notably, co-expression of AR and FOXA1 was observed by immunohistochemistry (IHC) in ~15% of triple-negative breast cancer (TNBC) patients24, and AR-positive (AR+), FOXA1-positive (FOXA1+) patients had a significant decrease in recurrence-free survival and overall survival compared to TNBC patients25. These findings suggest that when co-expressed in TNBC, AR, and FOXA1 may be mediating an estrogen-like gene signature similar to those expressed in luminal breast cancers. FOXA1 has been studied extensively in the context of ER chromatin binding, and ER binding is dependent on FOXA1 in the presence or absence of ligand23. Further, similar to findings in prostate cancer, 1.8% of breast cancers harbor mutations in FOXA1, and amplifications of the FOXA1 gene locus have been observed in breast and prostate cancers26. Notably most identified mutations are in the forkhead domain of FOXA1, and tumors in this study were exclusively ER+. The implications of these mutations, however, is still under investigation in breast cancer. Interestingly, differences exist between the function of FOXA1 in directing AR binding in breast versus prostate cancers, and future studies may investigate the varied roles of FOXA1 in directing AR binding in TNBC and prostate cancer, in addition to investigating the role of AR when co-expressed with ER. Current literature suggests, however, that regardless of tumor type, FOXA1 is an important cofactor for directing the transcriptional activity of AR.
AR and PTEN
Prostate cancer
Expression of AR with PTEN has also been investigated in prostate cancer. In prostate cancer patients, high AR expression with low PTEN expression is associated with poor clinical outcomes27. In prostate tumors, with loss of PTEN, there are decreased levels of AR signaling28. Inhibition of PI3K in these tumors results in increased levels of AR signaling through loss of human epidermal growth factor receptor 2 (HER2)-mediated feedback inhibition of AR28. A direct physical interaction between AR and PTEN in low passage LNCaP cells has been shown to inhibit nuclear translocation of AR resulting in an increase in degradation of AR protein29. A pilot study suggested that high expression of both AR and PTEN in patients with advanced prostate cancer was associated with a higher risk of relapse at 30 months after surgery (85.7% of high AR and PTEN expressing patients verses 16.6% in patients with low AR and PTEN expression)30. Further, combination therapy with both antiandrogen (bicalutamide) and PTEN induction was shown to reduce prostate-specific antigen (PSA) promoter activity compared to PTEN alone31. Sequencing of metastatic-CRPC (mCRPC) patients revealed that AR and PTEN are among the most commonly aberrant genes, along with the ETS family and TP5332. Therefore, these data suggest that both AR and PTEN may influence prostate tumor growth and progression.
Breast cancer
There are opposing findings when comparing AR and PTEN transcript expression in prostate verses breast cancer. In breast cancer, there is an AR-binding motif located in the PTEN promoter, and there is a positive correlation between AR and PTEN transcript levels27. In addition, high expression of AR and PTEN is correlated with better clinical outcomes for breast cancer patients27. Interestingly, in AR + TNBC, AR interacts at an ARE located in the promoter of ERβ33, and ERβ also plays a role in regulation of PTEN expression to control tumor growth34. The interaction between AR and PTEN may be context specific and important for predicting outcomes for patients with AR+ disease: where AR expression is associated with disease progression in prostate cancer7, PTEN loss is also correlated with poor outcomes27,35,36. In breast cancer, however, loss of PTEN is also correlated with negative ER and PR status, and PTEN loss is associated with breast tumor progression37. Therefore, these results suggest that the function of PTEN may be context specific and understanding the nuances in situational signaling of AR may help elucidate the role for PTEN in AR+ disease progression.
Non-genomic AR functions
Prostate cancer
Prostate cancer cells exhibit rapid proliferation responses in response to androgen stimulation, suggesting non-genomic AR signaling. Upon activation with androgens or estrogens, cytoplasmic AR can activate MAPK/ERK signaling through an association with Src38. The activation of the Src/ERK pathway is dependent on androgen concentration (0.1–10 nM) and is inhibited at high concentrations (100 nM)39. Treatment with dihydrotestosterone (DHT) also induces rapid ERK1/2 phosphorylation; however, MAPK activation can be blocked pharmacologically using a MEK inhibitor, suggesting AR is activating the Raf1-MEK pathway resulting in MAPK activation40. Further, AR can also activate the phosphatidyl-inositol 3-kinase (PI3K)/Akt pathway leading to activation of mammalian target of rapamycin (mTOR)41. In addition, androgens can interact at the plasma membrane which is associated with the modulation of intracellular calcium and cAMP levels41,42. Many membrane-bound G-protein coupled receptors are also responsive to androgen treatment, leading to an increase in apoptosis43, phosphorylation of ERK44, or reduced cell migration and metastasis45. Together, these findings suggest that AR may also function within the cytoplasm or at the membrane to activate non-genomic functions.
Breast cancer
Similar to non-genomic AR functions in prostate cancer, the cytoplasmic roles of AR have also been investigated in breast cancer. Chia et al. demonstrated that AR is necessary and sufficient for ERK phosphorylation following DHT stimulation in MDA-MB-453 and HCC-1954 cells46. Further, inhibition of AR resulted in decreased levels of phospho-Elk1, phospho-RSK, and c-FOS in xenograft tumors and in patient tumors, corresponding to a decrease in ERK target proteins46. In TNBC, AR inhibition has also been shown to modulate the activity of the Ca2+-activated K+ channel, KCa1.1, which is associated with breast cancer invasion and metastasis47,48. Multiple groups have also studied the role of cytoplasmic AR phosphorylation49,50; however, additional work is required to understand how AR modifications influence cellular function and localization. At the membrane, many receptors mediate rapid responses to androgen signaling, representing novel membrane-ARs51,52. These signals, however, are complex as agonistic verses antagonistic effects are dependent on receptor stoichiometry52. Furthermore, AR is expressed in fibrosarcoma cells; however, a significant portion of AR is transcriptionally incompetent and does not bind to AREs upon activation. Rather, there is crosstalk between EGFR and AR, and treatment with bicalutamide decreases xenograft tumor growth53. Together these data from multiple cancer models suggest that AR has non-genomic functions affecting tumor growth both in prostate and breast cancer which warrant further investigation.
Cell growth and proliferation
Androgens and AR splice variants
Prostate cancer
A number of AR splice variants have been identified, and they play an important role in the development of CRPC. The gene encoding AR is located on the X chromosome, encoding nine exons that produce the full-length AR transcript54. Aberrant splicing of AR pre-mRNA, however, can result in the production of AR isoforms that are constitutively active. These isoforms can drive an AR transcriptional program even in the absence of androgen signaling, resulting in androgen independent tumor growth55,56. AR variants (ARVs) are present both in prostate cancer and breast cancer, and these variants commonly are truncated or have mutations in the AR LBD57. In addition, AR transcripts can have aberrant splicing, resulting in skipped exons58. ChIP studies have demonstrated that splice variants, including AR-V7, are able to bind canonical AREs as well as unique regions of additional genes58. AR splice variants have been shown to require expression of full-length AR (AR-FL) suggesting that a balance between ARV and AR-FL expression is required for resistance in prostate cancer models59.
The most common splice variant, AR-V7, lacks an LBD60. Clinically, in a cohort of prostate cancer patients treated with enzalutamide or abiraterone acetate, 39 patients (19%) had detectable levels of AR-V7 in circulating tumor cells (CTC)61. Patients with AR-V7 expression had lower PSA response rates and worse survival compared to AR-V7 negative patients61. In addition to reliance on AR-splice variants, resistance to ADT is also mediated through signaling of additional hormone receptors. The GR has been shown to be increasingly present in androgen-deprived prostate cancer patients (78% vs. 38% of untreated patients)62, and expression of GR is increased in xenografts that are resistant to ARN-509 (apalutamide)63. In addition, there is overlap between AR and GR binding at classic response elements as well as regulation by both DHT and dexamethasone, a GR agonist63. In prostate cancer cells, stimulation with dexamethasone in the presence of enzalutamide resulted in expression of AR target genes63, providing further evidence that GR signaling could compensate for AR in the presence of AR-antagonists.
Breast cancer
There are significantly fewer AR mutations observed in TNBC compared to CRPC; however, AR splice variants are still common. In breast cancer, AR-V7 is most highly expressed splice variant in basal tumors compared to other tumor types, with the lowest expression in luminal tumors60. Little is known about how AR-V7 may be contributing to antiandrogen resistance in AR+ TNBC or if it is functioning similarly to its observed role in CRPC64. In HER2-enriched patients, however, high AR-V7 expression is associated with significantly higher metastasis-free survival and disease-specific survival60. Therefore, the ability of a tumor to produce its own androgens, as well as its reliance on splice variants may also play an important role in understanding how AR is functioning to drive tumor growth in the context of ADT or antiandrogen therapies.
Importantly, differences also exist in preclinical cell lines used to study AR+ breast cancers. While common cell lines, including MDA-MB-453, MDA-MB-231, ZR-75-1, MFM-223, MCF-7, and T47D, have varying levels of AR-FL expression, ARV expression also varies widely among cell lines—both in total ARV expression and expression of specific ARVs65. Notably, MDA-MB-453 cells contain the AR-Q865H variant which harbors a mutation in the AR LBD66, demonstrating the importance of considering the influence of ARV expression in laboratory studies. Furthermore, understanding the similarities and differences of how ARVs may be influencing AR expression and contributing to breast tumorigenesis will be important.
Estrogen influence on androgen signaling in breast cancer
AR+, ER+ cancers
Breast tumors that are ER+ are more likely to be AR+ compared to tumors that are ER−67, and AR status is related to ER and PR status but independent of the status of HER268. Interestingly, patients with AR:ER ratios ≥2 have worse disease-free survival compared to patients with lower AR:ER ratios in the presence of antiestrogen therapies or chemotherapy treatment69. Defining expression of AR and ER, however, is challenging, and results vary widely depending on the assays (including IHC, radioimmunoassay, and reverse-phase protein array67) and cut-offs used to define positivity. Clinically, ER expression is measured by IHC, and ER+ tumors are defined as those with >1% of tumor cells with positive nuclei70. AR positivity, however, has been defined with varying cut-off levels from 1 to 75%67,71. The prognostic role of AR in breast cancer remains unclear. A recent study has demonstrated that >78% AR positivity is required to accurately assess the prognostic role of AR in ER+ cancers, with ER+ patients that have ≥78% AR positivity having the best survival outcomes72. In other studies, however, breast cancer patients with AR+ tumors have better overall survival at both 3 and 5 years compared to patients with AR− tumors, regardless of ER expression67. These data suggest that the role of AR in driving breast cancer growth may differ in the presence or absence of ER and that antagonizing AR may have different effects depending on the level of AR expression.
AR expression in ER+ breast cancers antagonizes the signaling of mitogenic ERα, and AR expression leads to the upregulation of ERβ33. In ER+ breast cancer, AR binds at an ARE located in the promoter of the ERβ gene, resulting in increased ERβ expression33. Interestingly, the presence of ERβ has been shown to inhibit transcriptional activity of ERα73, therefore, suggesting that AR-regulated increased activity of ERβ may indirectly influence ERα activity. Similarly, in prostate tissue, ERβ is thought to play an antagonistic role to AR, resulting in the suppression of cellular proliferation and the promotion of apoptosis74. ERβ is also important for the control of cell cycle progression and arrest75,76, indicating that increasing ERβ expression may be a therapeutic strategy in prostate cancer77.
In contrast to early studies suggesting high AR expression is associated with improved outcomes, recent data suggest high AR expression may be associated with therapy resistance, including endocrine therapy resistance. Indeed, De Amicis et al. first reported the positive correlation between high AR expression and tamoxifen resistance, suggesting that tumors with a high AR:ER ratio are more likely to be resistant to antiestrogen therapies, which are common first line of therapy for ER+ tumors78. Patients resistant to tamoxifen with AR:ERα ratios ≥2 have worse disease-free survival, and disease-specific survival79. Interestingly, in tamoxifen-resistant MCF-7 cells, loss of AR signaling by AR knockdown, but not treatment with enzalutamide, restored sensitivity to tamoxifen80. These results suggest that AR expression may be a mechanism of hormone therapy resistance, and therefore a therapeutic target in resistant hormone receptor positive breast cancers.
Anti-AR therapy is of increasing clinical interest. AR inhibition may be an effective strategy for growth inhibition of AR+, ER+ breast tumors. AR inhibition with enzalutamide has been shown to be synergistic with tamoxifen- or fulvestrant-mediated ER inhibition, in addition to controlling growth of tamoxifen-resistant MCF-7 cells in vitro and in vivo in an AR+, ER+ patient-derived xenograft model81. Enzalutamide has been shown to be effective in AR+ breast tumors, including ER+ (MCF-7) cells and ER− (MDA-MB-453) cells82. ChIP analyses demonstrate that there is extensive overlap between AR and ER binding sites after E2 stimulation in MCF-7 cells81. Interestingly, however, AR binding was different based on stimulation with DHT or E2 in MCF-7 cells suggesting that AR may regulate a unique transcriptional program in the absence of estrogen signaling, providing additional evidence for synergism between antiestrogen and antiandrogen therapies81. These results indicate that targeting AR in combination with anti-ER therapies may be an effective therapeutic strategy for patients with AR+, ER+ breast cancers.
Functionally, AR and ER share many similarities in their signaling pathways, including the mechanism of receptor activation, as both receptors are activated through ligand binding83. ER and AR recognize similar sequences of DNA: where ER binds to 5'-AGGTCA-3', AR recognizes the 5'-AGAACA-3' sequence83,84. Further, in breast cancer, both AR and ER require similar cofactors for the activation of common signaling pathways83. Binding of AR or ER can activate MAPK signaling, among other pathways83, and due to their similar structure and signaling function, both hormone receptors are in competition within the cell for the binding of scaffold proteins and cofactors83. While AR and ER share many similarities, there may be important differences determining their role in driving tumor growth.
AR+, ER− cancer
The function of AR in breast cancer appears to be dependent upon its co-expression with ER, as there is evidence for varying effects of AR on the growth of breast cancer cells in the presence or absence of ER. Indeed, while AR is co-expressed with ER in 70–90% of breast tumors, AR is only expressed in 15–30% of ER-negative breast tumors85. Breast cancers that do not express ER, PR, and HER2 have been traditionally described as TNBC. Recently, however, a subtype of TNBC has been established which is characterized by luminal AR expression86,87. In studies with AR+ human breast cancer cell lines, androgens had both proliferative and antiproliferative effects depending on the cell line of interest88. More recently, however, multiple groups have demonstrated that targeting AR in AR+ TNBC is an effective treatment strategy both in vitro and in vivo82,89,90. Interestingly, in AR+ TNBC, ~30% of patients have expression of ERβ91, and ERβ expression has been shown to increase the efficacy of antiandrogens in AR+ TNBC cells92. Together these data demonstrate the importance of AR in driving growth of AR+ TNBCs.
While AR expression has been increasingly recognized in AR+, ER− breast cancers, the specific role of AR signaling is not well understood. Some studies suggest an important role for AR in signaling in the absence of ER93. In an analysis of AR+, ER− MDA-MB-453 cells, the AR cistrome was found to be more similar to that of ER in MCF-7 (AR−/ER+) cells compared to the AR cistrome in LNCaP prostate cancer cells22. Therefore, AR may function in place of ER in AR+, ER− breast cancer22, having a distinct role in AR+ TNBC compared to prostate cancer. AR may also be important for promoting the cancer stem cell-like (CSC-like) population in TNBC, in addition to reducing the levels of detachment-induced apoptosis in cells grown in forced suspension compared to attachment conditions94. These results suggest that AR blockade may be effective in combination with paclitaxel to target CSC-like cells and reduce tumor recurrence compared to paclitaxel treatment alone94. In addition, AR is commonly enriched in breast cancers overexpressing HER2, indicating a role for AR in activation of HER2 and Wnt signaling89,95. Therefore, AR expression may be an important target for directing treatments for patients with ER- breast cancer.
DNA damage repair
Prostate cancer
While the mechanism of AR in response to DNA damage is just beginning to be uncovered in breast cancer, the mechanistic role of AR in DNA damage repair has been more extensively characterized in prostate cancer. Goodwin et al. found that AR is activated in response to reactive oxygen species (ROS) and DNA damage96. Additionally, in response to ionizing radiation, CRPC cells have enhanced DNA repair and decreased DNA damage97. AR activation results in the expression of DNA damage repair genes including PRKDC, encoding DNA-dependent protein kinase catalytic subunit (DNA-PKcs), an essential protein necessary for nonhomologous end joining (NHEJ) repair of double-stranded DNA (dsDNA) breaks96. In addition, treatment with radiation and androgens results in the upregulation of XRCC2 and XRCC3, two genes important for homologous recombination (HR)96. Conversely, antiandrogen treatment results in decreased DNA repair in cells and increased levels of dsDNA breaks97. The same group also showed that treatment with AR inhibitors results in increased radiosensitivity and decreased NHEJ-mediated recombination suggesting that AR is involved in NHEJ-mediated repair of dsDNA breaks97. DNA-PKcs has been shown to function in complex with Ku70 and Ku80 to respond to DNA damage. Interestingly, DNA-PKcs physically interacts with AR; however, this interaction does not require the presence of DNA98. This suggests that AR regulation of the DNA damage response may not be completely dependent on AR-mediated transcriptional regulation of DNA damage response genes. Following androgen stimulation in prostate cancer cells, AR is recruited to enhancer elements, along with DNA-PKcs, coregulator p300, and RNA Pol II suggesting that the interaction of AR and DNA-PKcs may be important for the regulation of specific transcriptional programming98. Therefore, an interaction between AR and DNA-PKcs may also be important for AR’s role in the repair of DNA damage. In patient tissue, castration resulted in the downregulation of Ku70 protein levels, impairing NHEJ99. AR regulates Ku70 levels in prostate tissue, and due to the critical role of Ku70 in effective NHEJ, downregulation of this protein abrogates NHEJ-mediated repair99. Collectively these data suggest that AR signaling plays an important role in the repair of dsDNA breaks, at least in part through interactions with Ku70/Ku80 and DNA-PKcs, members of the NHEJ repair pathway.
Breast cancer
Recent data in breast cancer suggest that loss of AR signaling through knockdown or pharmacologic inhibition with enzalutamide or seviteronel results in increased sensitivity to ionizing radiation100,101. In addition, AR mRNA levels correlate with survival following radiation treatment, and AR is important for regulating the DNA damage response in AR+ breast cancer cell lines102. Pharmacologic AR inhibition results in delayed repair of dsDNA breaks following ionizing radiation, suggesting that AR is influencing dsDNA damage repair. Additionally, AR inhibition with enzalutamide decreases levels of phosphorylated DNA-PKcs following radiation, indicating that NHEJ may be important for the repair of radiation-induced dsDNA breaks in breast cancer100. Although some similarities exist between the role of AR in DNA damage repair in prostate and breast cancers, a full characterization of the similarities and differences is still ongoing.
Cell cycle regulation
Prostate cancer
Cell cycle progression is driven by the rising and falling in levels of cyclins and cyclin dependent kinase (CDKs), in addition to the activation of these proteins103. In prostate cancer, AR is regulated in a cell-cycle-dependent manner104. Nuclear transactivation of AR is highest in G1 and decreases in S phase, while the same changes occur in AR phosphorylation and cellular localization104. Further, CDK1 has been shown to phosphorylate AR on S308 in response to ligand binding104. The phosphorylation results in changes in AR chromatin localization104. AR signaling is responsible for the activation of genes controlling the G1–S transition105. Specifically, AR is responsible for G1 CDK activation and the phosphorylation of retinoblastoma (pRb), which is necessary for the activation of CDKs that will drive the G1–S phase progression105. In the absence of androgen signaling, prostate cancer cells will arrest in early G1 phase as they do not have expression of the necessary CDK and cyclin proteins106,107. AR and pRb have also been shown to interact, and an overexpression of pRb increases the transcriptional activity of AR108,109.
AR signaling is also important for the regulation of other cell cycle related genes, including the regulation of CCND1 expression110. Importantly, CCND1 encodes cyclin D1 which has an interaction with pRb that is necessary for cell cycle progression. AR binds to AREs that are located ~570–556 base pairs upstream of the transcription start site of the proximal promoter of CCND1, suggesting that AR plays a regulatory role to influence CCND1 expression110. In prostate cancer cells, following treatment with androgens, there is induction of expression of CDK inhibitors p21 and p27111. Expression of p21 is controlled at the transcriptional level through the presence of an ARE in the promoter region, ~200 base pairs upstream of the proximal promoter112. AR signaling has been shown to be important for control of cell cycle-related gene expression, resulting in growth implications in tumor cells.
Additionally, in prostate cancer cells, the synthetic androgen mibolerone inhibited proliferation and reduced levels of c-MYC transcripts, suggesting that AR is important for regulating c-MYC levels113. AR expression is also regulated by AREs as well as MYC binding at the consensus site114. Thus, in addition to its role cell growth and the DNA damage response, AR expression and activation is itself regulated in a cell cycle-dependent manner which then influences expression of CDK and transcription factors to regulate progression through the cell cycle.
Breast cancer
In addition to interactions with cyclins and CDKs, AR also interacts with many other important proteins, including well characterized oncogenes and tumor suppressers. In AR+ TNBC, DHT has been shown to increase levels of cyclin D1, while decreasing p73 and p21 expression115. Conversely, treatment with bicalutamide resulted in a decrease in cyclin D1 expression, while increasing p73 and p21 levels, implicating a role for AR in the control of cell cycle progression in AR+ TNBC models115. The expression of AR and pRb in breast cancer is also significantly correlated, and AR has been shown to interact with other transcription factors, including MYC, which are important for cell cycle control116. In breast tumors, high AR expression is negatively correlated with MYC overexpression116. MYC expression has been linked to cell proliferation, and inactivation of MYC impairs cell cycle progression as MYC targets cell cycle regulators like cyclins, CDKs, and E2F transcription factors117. Additionally, in breast cancer models, the presence of an ARE −383 to −377 base pairs upstream of the ERβ promoter region results in enhanced control of ERβ expression as a result of AR signaling33. ERβ has been shown to negatively regulate transcription of c-MYC, cyclin D1, and cyclin A, while also increasing transcription of CDK inhibitors like p21 and p2733. In ER+ breast cancer models, DHT-mediated activation of AR has been shown to inhibit ERα signaling and cell cycle progression through a reduction in cyclin D1 transcription118. Further, AR and ER both require the steroid receptor coactivator AIB1118 which is commonly expressed in breast cancers119,120, and high AIB1 expression is correlated with poor mortality121. Therefore, through direct or indirect mechanisms, AR signaling likely also plays an important role in controlling cell cycle progression in breast cancer.
Metastasis
Prostate cancer
AR has been shown to contribute to the formation of metastases. The AR pathway and AR splice variants have been implicated in metastatic phenotypes in prostate cancer122. Gene array and IHC data of both primary and metastatic tumors demonstrate that AR mRNA and protein expression are significantly higher in metastases compared to primary prostate lesions123. In vitro, increased AR expression in prostate tumors also led to the formation of metastases and induction of the epithelial to mesenchymal transition (EMT)123, the process by which cells lose their polarity and gain the ability to migrate and become invasive. In addition, during prostate cancer development, the presence of fibroblasts provides important structural and functional changes that regulate the extracellular matrix124. Expression of nuclear receptors has been shown to be important in squamous cell carcinoma cancer-associated fibroblasts (CAFs) compared to normal-associated fibroblasts, with nuclear receptors influencing many cellular functions including invasiveness125. Additionally, AR expression in prostate CAFs has been shown to promote growth and invasion126. AR activation in the stroma has been shown to be essential for prostate cancer progression and metastasis127. Interestingly, the AR cistrome in prostate CAFs is distinct from the AR cistrome in epithelial cells suggesting a novel role for AR in the microenvironment128. Notably, AR relies on AP-1 in the stroma, rather than FOXA1 as observed in epithelial cells128.
Furthermore, the regulatory role of AR in gene expression has been shown to be important for the regulation of prostate cancer metastases. In this context, AR negatively regulates expression of ZBTB46, a tumor promoter through miR-1129. Therefore, disruption of AR signaling can result in overexpression of ZBTB46 resulting in an increase in transcriptional regulation of SNAI1, a driver of EMT, resulting in metastasis formation129. Further, AR inhibition with enzalutamide has been shown to increase metastases by decreasing EPHB6 suppression leading to JNK signaling resulting in cell invasion130. These findings suggest that AR plays an important role in controlling metastatic progression of prostate tumors, demonstrating the importance of future work in this area.
Breast cancer
In patients with breast cancer, metastases are likely to have multiple drivers of disease progression. In preclinical models, AR has also been shown to contribute to invasiveness and migration of TNBC cells through activation of the Src complex131. When MCF-7 cells were treated with DHT, there was also an increase in invasion and migration, as well as a decrease in epithelial markers and an increase in mesenchymal markers132. DHT treatment also induced other markers of EMT suggesting that AR activation may promote EMT in MCF-7 cells132.
As with prostate cancer, previous data from breast cancer patients demonstrates that AR expression is conserved from the primary tumor into metastases133,134. One study suggests that there is 78.6% agreement in AR status in primary tumor and lymph node metastases135. In the discordant cases, 60/72 had AR- primary tumors, and AR+ lymph node metastases135. Further, IHC analyses in tumors and metastases showed greater than 60% agreement between the expression of AR in primary tumor and metastases136.
Treatments targeting androgens and the AR for prostate and breast cancer
Pharmacological agents have been developed to inhibit AR binding to androgens and AR activation due to its role in driving cancer development and progression (Fig. 1). Many of these agents have been effective in the treatment of prostate cancer, and the clinical applications have been expanded to women with AR+ breast cancers. Here we explore these various agents, their mechanisms of action, and the data that exist in the treatment with women with breast cancer, including the ongoing clinical trials assessing their use in women with AR-positive breast cancer and the emerging results from these trials (Tables 1 and 2).

Androgens like testosterone (T) are produced from cholesterol. CYP17-lyase inhibitors and aromatase inhibitors (1), including abiraterone acetate, seviteronel, CR1447, orteronel, galeterone, block the conversion of cholesterol into testosterone. Additionally, luteinizing hormone-releasing hormone (LHRH) or gonadotrophin-releasing hormone (GnRH) antagonists function to reduce levels of circulating androgens, and ketoconazole inhibits production of testosterone. When androgens enter the cell, they can be converted to DHT, a more potent AR agonist. This reaction requires 5α-reductase, an enzyme that can be inhibited by finasteride and dutasteride (5). Antiandrogens (2), including flutamide, bicalutamide, enzalutamide, apalutamide, darolutamide, CR1447, seviteronel, and TRC253, block androgen binding to AR or inhibit AR function. Many antiandrogens, including enzalutamide, apalutamide and darolutamide, inhibit nuclear translation of AR, preventing DNA-binding and downstream gene transcription. Antisense oligonucleotides bind to mRNA encoding AR, preventing protein translation (4). AR degraders (6) promote protein ubiquitination and proteasomal-mediated degradation to lower AR protein levels intracellularly.
Androgen deprivation therapy
The use of ADT is universally accepted as a first line therapy for metastatic prostate cancer137. This treatment attempts to lower levels of serum testosterone in men with prostate cancer to prevent tumor growth138. This is done chemically with the use of luteinizing hormone-releasing hormone (LHRH) or gonadotrophin-releasing hormone (GnRH) antagonists, like Degarelix, Goserelin, and Leuprolide, which are used to suppress the production of androgens137. Many clinical trials also are assessing the efficacy of ADT in combination with other treatment strategies in an attempt to improve ADT efficacy, especially in cases where AR mutations cause castration to be ineffective at controlling disease progression.
5α-reductase inhibitors
5α-DHT is produced from testosterone in specific tissues, including the prostate, through the enzymatic activity of 5α-reductase. Compared to testosterone, DHT has a slower dissociation rate from AR, suggesting that AR-DHT is a more stable complex, making DHT the preferred AR ligand139,140. Competitive inhibitors of 5α-reductase, like finasteride or dutasteride, can be used to lower levels of serum and prostate DHT141,142,143,144. The effects of these 5α-reductase inhibitors, however, are complex as they may not exclusively target the enzymatic activity of 5α-reductase and likely have additional off-target AR inhibitory effects as well145.
CYP17-lyase inhibitors
Abiraterone acetate is a selective inhibitor of cytochrome P450 17alpha-hydroxylase/17,20-lyase (CYP17), which, through its function, decreases the adrenal and tumoral synthesis of androgens146. CYP17-lyase inhibitors lower androgen availability to reduce the activation of androgen signaling. Trials in men with chemotherapy-naive CRPC concluded that treatment with abiraterone acetate and prednisone prolongs overall survival compared to treatment with prednisone alone (NCT00887198)147. In a phase II trial for women with triple negative, AR+ locally advanced or metastatic breast cancer (NCT01842321), treatment with abiraterone acetate and prednisone also provided benefit for some patients148. Of 138 patients assessed for the trial, 53 (37.6%) had AR+ TNBC: 34 of these patients were included. This trial assessed the clinical benefit rate (CBR) for 30 of the patients at 6 months with a CBR of 20.0% (95% CI: 7.7–38.6%) including one patient who had a complete response (CR) and 5 patients with stable disease (SD) at ≥6 months148. Secondary outcomes included objective response rate (6.7%, 95% CI: 0.8–22.1%), and progression-free survival (median time: 2.8 months, 95% CI: 1.7–5.4)148. These studies suggest that treatment with abiraterone acetate may be a beneficial treatment strategy for both men with CRPC and women with molecular apocrine breast cancer.
Other CYP17-lyase inhibitors include galeterone (TOK-001) and orteronel (TAK-700)149. Galeterone has been shown to be effective in reduction of PSA levels and was well-tolerated by patients in early clinical trials150. Orteronel treatment is effective at suppressing testosterone levels and shrinking the androgen-dependent organs including the prostate gland151. Phase III clinical trials found that orteronel and prednisone treatment verses placebo and prednisone gave patients longer progression-free survival (PFS); however, men with orteronel and prednisone treatment did not have extended overall survival152. In breast cancer, there are currently phase I and II clinical trials assessing the use of orteronel in patients with metastatic breast cancers that express AR (NCT01808040, NCT01990209). NCT01808040 assesses the safety of orteronel use for the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer in addition to measuring the estradiol levels in these patients following treatment153. NCT01990209 is a phase II trial for male or female patients with metastatic AR+ BC (TNBC or ER+ and/or PR+ BC) with primary outcome measures of response and disease control rates. This trial will also assess safety, PFS, OS, and serum hormone levels in addition to screening tumors for PTEN expression and PIK3CA mutations. Due to failure in phase III clinical trials in men with prostate cancer, orteronel was taken out of development in 2014154.
Antiandrogens
Antiandrogens are a class of agents which act as nonsteroidal competitive inhibitors of the AR155. Flutamide and bicalutamide are two such agents that have been used to block androgen binding and abrogate nuclear AR signaling. Although AR targeting has been a strategy for over 30 years, original phase II clinical trials with flutamide suggested it did not have antitumor activity which delayed the initiation of further trials with the drug156. Recent studies, however, have shown that flutamide treatment is effective and well-tolerated for treating PSA recurrence following prostatectomy, radiation therapy, or cryotherapy for patients with prostate cancer157. In addition, in breast cancer, bicalutamide has been shown to have a CBR of 19% in patients with AR+, ER−, PR− metastatic breast cancer where 12% of tumors were AR+158. These results suggest that antiandrogen therapies are effective for the treatment of patients with traditionally hormone receptor-negative breast cancers. Unfortunately, in prostate cancer, it has been shown that exposure to antiandrogens can augment frequency of AR mutations and variants159, and metabolites of antiandrogens can result in stimulation of prostate cancer cell growth as flutamide metabolites function as an AR agonist160. There are additional ongoing clinical trials that are assessing the use of flutamide as a second line treatment of patients with CRPC who have relapsed after ADT and bicalutamide treatment (NCT02918968) or using flutamide treatment to prevent prostate cancer in patients with neoplasia of the prostate (NCT00006214). In addition, NCT02910050 is investigating the use of bicalutamide with AIs in AR+, ER+ breast cancers161.
Second generation antiandrogens
Four FDA-approved second generation antiandrogens, abiraterone acetate, apalutamide, darolutamide, and enzalutamide, improve upon the first-generation antiandrogens. Enzalutamide is able to inhibit the growth of both ER+ and ER− breast tumors by inhibiting AR nuclear translocation146. In addition to growth inhibition, enzalutamide also can inhibit tumor cell migration and invasion162. In mCRPC patients who had previously received chemotherapy treatment, treatment with enzalutamide also contributed to prolonged survival (NCT00974311)163. In breast cancer, a Phase II trial (NCT01889238) for women with advanced, AR+ TNBC tested the use of enzalutamide for improving outcomes and CBR for patients at 16 weeks (CBR16) as well as assessing clinical benefit at 24 weeks (CBR24), PFS, response rates, and safety of enzalutamide treatment164. This study also found that 47% of the 118 enrolled patients had an AR related gene signature, and clinical outcomes were better for patients with AR+ disease164. No new side effects were reported from enzalutamide treatment in this trial, indicating its potential use as a therapeutic for women with TNBC164.
Apalutamide (ARN-509) is a second generation AR antagonist, similar to enzalutamide, that binds to the LBD of AR to inhibit nuclear translocation and ARE binding149. Apalutamide has a seven- to ten-fold increased binding affinity to AR compared to bicalutamide165. In preclinical studies, apalutamide had antitumor activity in a castration-sensitive model of prostate cancer166. There were also lower levels of apalutamide in mouse steady-state plasma and brain levels compared to enzalutamide treatment, which could indicate lower frequency of seizures with apalutamide166. In preclinical studies, apalutamide also had antitumor and growth inhibitory effects in AR+ TNBC cells166. Results from the SPARTAN trial, a Phase III clinical trial (NCT01946204) for men with nonmetastatic castration-resistant prostate cancer (nmCRPC), demonstrated improved metastasis-free survival in patients treated with apalutamide compared to placebo167. Following this trial, apalutamide was approved by the FDA for treatment of nmCRPC165. To date, there have been no trials with apalutamide in patients with AR+ breast cancer.
Darolutamide (ODM-201) is an AR inhibitor that binds wild-type AR with a higher affinity than enzalutamide to block AR nuclear translocation149. In addition, darolutamide can be effective against mutant ARVs which can develop with resistance to enzalutamide and apalutamide therapy168. In prostate models, darolutamide has low brain-penetrance and treatment does not produce an increase in mouse serum testosterone levels168. Recently, results from the ARAMIS trial (NCT02200614), a phase III trial for nmCRPC patients, demonstrate that darolutamide provides better metastasis-free survival compared to placebo169. The START trial is a phase II trial for women with AR+ TNBC comparing darolutamide treatment with capecitabine, an antimetabolite chemotherapeutic (NCT03383679). This trial investigates CBR16 as a primary objective, and CBR24, response rates, overall survival, PFS, and safety as secondary objectives for women with locally recurrent or metastatic AR+ TNBC.
Novel compounds
A number of novel compounds have also been developed to block or abrogate androgen signaling. Seviteronel (VT-464) is a nonsteroidal selective CYP17-lyase inhibitor and AR antagonist that both blocks testosterone and estrogen production and inhibits AR activation170, rendering it a potentially effective alternative to agents which either inhibit androgen production or AR activation. Clinical trials for patients with ER+ or TNBC indicated that seviteronel was well-tolerated in women, with the majority of adverse events (AEs) being Grade 1/2, in addition to four Grade 3/4 AEs that may be related to seviteronel treatment171. Phase I trials in CRPC patients suggest that seviteronel may be an effective treatment alternative for men who are not responsive on other therapies with most reported AEs being Grade 1/2172. Preclinical work in AR+ TNBC demonstrates that seviteronel inhibits cell proliferation and growth on soft agar173. ChIP-seq and RNA-seq analyses demonstrate that AR-regulated genes are increased with DHT stimulation and decreased in mice treated with seviteronel173. Trials with seviteronel continue to be ongoing for patients with CRPC, AR+ TNBC, or men with ER+ breast cancer, who had previously been treated with enzalutamide (NCT02130700, NCT02012920, NCT02580448, NCT03600467). The CLARITY-01 trial (NCT02580448) is assessing the CBR at 16 or 24 weeks for women with ER+ or TNBC or men with locally advanced or metastatic breast cancer who are receiving seviteronel treatment174. Of the patients enrolled for stage 1, CBR16 for TNBC patients was 2 of 6, and CBR24 for ER+ patients was 2 of 11174. Of patients with CTCs, 7 of 10 had a CTC decline at C2D1174. Patients receiving seviteronel also had a decline from baseline in concentrations of estradiol and testosterone175. The most common AEs were tremor, pain, fatigue and dyspnea, nausea, AST increase, ALT increase and abdominal pain, suggesting that seviteronel was well-tolerated175. These results indicate that seviteronel may be a potential therapeutic option for the treatment of AR+ disease.
CR1447 (4-hydroxytestosterone [4-OHT]) is a novel AR inhibitor that acts both as a steroidal AI as well as an AR antagonist by binding to AR176. When injected, 4-OHT is converted to 4-hydroxyandrostenedione (4-OHA), a previously used form of AI that was injected for the treatment of breast cancer176. Both 4-OHT and 4-OHA are unable to be made into estrogens in vivo176. Preclinically, CR1447 has been shown to inhibit growth of AR+ BC cell lines, but not AR knockout cell lines, or those with siRNA-mediated AR knockdown176. Results from a Phase I clinical trial (NCT02067741) indicate that, when topically administered, CR1447 was well-tolerated with grade 1/2 AEs and no dose-limiting toxicities (DLTs) in 12 patients with ER+/HER2− breast cancer176. Two patients (17%) had stable disease after 12 weeks of treatment176. Therefore, CR1447 may also be viable treatment option.
Enobosarm is a selective androgen receptor modulator (SARM) that was originally tested in Phase I, II, and III clinical trials for its use in improving lean body mass and treating cachexia177. Enobosarm has tissue specific activity, with anabolic activity in muscles and bone without affecting growth of hair in women and prostate in men178. It has been well-tolerated by both men and women; additionally, in patients with advanced cancer, treatment with enobosarm leads to an increase in lean body mass179. Enobosarm has also been well-tolerated as an androgen agonist in women with AR+ metastatic breast cancer180. Androgen-based AR agonists have previously been shown to be effective for the treatment of breast cancer181, and enobosarm similarly stimulates AR, but unlike androgens, does not have masculinizing side effects182. A phase II trial (NCT01616758) assesses CBR, and PSA is evaluated as a biomarker of AR activity. In addition, NCT02971761 is investigating the use of pembrolizumab with enobosarm for AR+ TNBC patients183. Enobosarm may soon join the treatment armamentarium.
Antisense oligonucleotides (ASOs) have also been used to inhibit AR-driven gene expression, especially in contexts where AR is activated independent of hormone binding. ASOs bind to mRNA, causing the mRNA to be degraded, therefore reducing levels available for protein synthesis. Prostate cancer models have shown that ASOs are able to reduce AR expression, resulting in decreased cell growth184,185. In addition, ASOs used against AR mRNA were able to shut down the downstream activation of AR-mediated genes in hormone-independent conditions186. ASO administration in mouse models did not have any observed side effects and, compared to castration, did not result in shrinking of mouse prostates185. Use of ASOs may also be a method for targeting AR splice variants as two ASOs have been used to effectively silence AR-V7, but not AR-FL, signaling in CRPC cell lines187. Therefore, these findings suggest that the use of ASOs may be a useful strategy for overcoming the resistance that often develops to antiandrogens in prostate cancer. In addition, ASOs may also be an effective treatment strategy for targeting mutant ARVs.
Targeted degradation of proteins with the use of Proteolysis Targeting Chimeras (PROTACs) is a novel method for the inhibition of AR signaling in prostate cancer cell models. PROTAC-mediated degradation takes advantage of E3 ubiquitin ligase activity by linking a ligand for the target protein to a ligand for the E3 ubiquitin ligase188. Upon ligand binding to the protein of interest, the protein is ubiquitinated by the E3 ubiquitin ligase resulting in degradation by the 26S proteasome. Multiple AR degraders have been developed using PROTAC for use in prostate cancer189,190, and they have been shown to be more effective than enzalutamide in vitro and in vivo in models of enzalutamide-sensitive and resistant prostate cancer191,192. Enhanced efficacy of AR degraders in prostate cancer models may demonstrate the importance of removing AR protein as opposed to pharmacologically inhibiting AR activity for the treatment of resistant prostate cancers. In the future, pharmacologic AR degraders may be introduced clinically for the treatment of aggressive AR-driven cancers.
There are also additional compounds that have limited use in treating AR-driven disease. Ketoconazole is an antifungal agent that is also able to competitively bind to the AR193. Ketoconazole has also been shown to inhibit enzymes important for testosterone synthesis194 and is under investigation in combination with docetaxel (NCT00212095)195. In addition, TRC253, a novel competitive inhibitor of AR has been shown to be an antagonist to wild-type AR as well as all tested AR mutants196, including AR F877L, a mutation occurring in the LBD of AR197.
Combination therapies
AR + radiation therapy
Prostate cancer
Radiotherapy has been shown to induce AR expression in prostate cancer cells, and ADT sensitizes cancer cells to radiotherapy198. Treatment with enzalutamide was also shown to radiosensitize prostate cancer cells more effectively than ADT199. Combination treatment with enzalutamide and radiation therapy resulted in a significant increase in apoptosis and senescence compared to treatment with enzalutamide or radiation alone199. In prostate cancer, radiosensitization was also observed with ARN-50997. In addition, treatment with antiandrogen therapies resulted in the downregulation of DNA repair genes, thereby promoting radiosensitivity through a decrease in NHEJ activity97.
Breast cancer
The AR has been shown to be a potential mediator of radioresistance and a target for the radiosensitization of AR+ TNBC100,101,102,200. Inhibition of AR with enzalutamide results in increased radiosensitization of AR+ breast cancer cells through the inhibition of AR-activated DNA-PKcs-mediated repair100. Similar results were observed with seviteronel, the dual CYP17 inhibitor and AR antagonist101, however the differences in the mechanisms of radiosensitization with these agents need to be further assessed.
AR + PARP inhibitors
Prostate cancer
Poly ADP-ribose polymerase (PARP) is a nuclear enzyme that modifies substrates through the addition of PAR moieties201. Cancers with mutations to BRCA1 or BRCA2 have HR deficiencies, rendering them increasingly susceptible to treatment with PARP inhibitors. Inhibition of PARP in tumors with BRCA mutations results in synthetic lethality and forces cells to rely on NHEJ for repair of DNA breaks. PARP has been shown to be recruited to sites of AR binding and promotes AR function201. When AR is inhibited, HR deficiency and BRCAness is induced202. Therefore, AR activity is important for the maintenance of HR gene expression. Following ADT, PARP levels are elevated leading to prostate cancer cell survival203. Combination therapy of PARP inhibition with ADT may be important for the impairment of HR before the tumors become castration resistant203.
PARP also plays an important role in the AR signaling cascade. Combination treatment with the PARP inhibitor talazoparib with enzalutamide or abiraterone acetate has significant synergy204. Antiandrogen therapies induce PARP cleavage, resulting in an increase in dsDNA breaks204. This synergy is a therapeutic target for CRPC patients with mutations in DNA damage repair. Therefore, cancer cells with DNA damage repair mutations are more sensitive to PARP inhibitors due to the role of the AR in the transcriptional regulation of DDR genes205.
Breast cancer
PARPi has been established to be an effective treatment strategy for patients with breast cancers harboring mutant BRCA1 and BRCA2206. To date, the combination therapy of PARPi with anti-AR therapy has not been tested in breast cancer; however, this combination may be an effective treatment strategy for AR+ BC patients, especially those with BRCA mutated tumors. Because many PARPi can induce PARP trapping, resulting in the formation of dsDNA lesions207, and anti-AR therapies have been demonstrated to result in a delay in dsDNA break repair in the presence of DNA damage, combining these therapies may be effective in creating deleterious lesions for tumor cells. Future work may assess PARPi in combination with anti-AR therapies for the treatment of AR+ breast cancers.
AR + CDK4/6 inhibitors
Prostate cancer
AR regulates cell cycle progression through the G1–S phase transition, therefore promoting CDK activity and inducing phosphorylation for the inactivation of pRb105. Due to crosstalk of AR with CDK/pRb in promoting cell cycle progression, combined AR and CDK4/6 inhibition has also been shown to be a therapeutic strategy in prostate cancer208.
Breast cancer
Palbociclib, ribociclib, and abemaciclib are selective inhibitors of CDK4/6 and are widely used for the treatment for ER+ breast cancer. A Phase I/II clinical trial is currently assessing the use of palbociclib with bicalutamide for treatment of AR+ metastatic TNBC (NCT02605486). This trial will establish recommended doses for the combination therapy in addition to measuring PFS, and secondary outcomes including response rates, CBR, and safety209.
AR + PI3K inhibitors
Prostate and breast cancer
Phosphatindylinositol 3-kinase (PI3K) is an enzyme involved in cellular functions including cell growth, proliferation, and differentiation; however, PI3K is also highly mutated in cancer. Qi et al. found that inhibition of both AR and PI3K can be synergistic as AR and PI3K signaling work through reciprocal feedback loops210. Combined inhibition of AR with the PI3K or mTOR pathway suppressed cell proliferation and resulted in an increase in apoptosis and cell cycle arrest in CRPC cells210. An ongoing trial is investigating the treatment of taselisib, a PI3K inhibitor, and enzalutamide in patients with AR+ metastatic TNBC (NCT02457910). This trial will assess dose-limiting toxicities to determine the maximum tolerated dose in addition to measuring patient response and CBR211.
Phase III development of antiandrogen treatments
Many antiandrogen treatment strategies have been effectively translated from preclinical studies into clinical use through the use of clinical trials. For women with metastatic, AR+ TNBC, there is a phase III clinical trial (NCT03055312) underway comparing conventional chemotherapy to bicalutamide treatment. This trial will assess the CBR at 16 weeks as well as progression-free survival at 24 months. The ENDEAR trial (NCT02929576) is a phase III trial comparing PFS for patients treated with paclitaxel chemotherapy +/− enzalutamide or enzalutamide followed by paclitaxel treatment; however, this trial was withdrawn. Finally, there is an ongoing feasibility trial (NCT02750358) of enzalutamide in women with AR+ TNBC that should report preliminary DFS and OS data in the coming year212,213. Data recently presented from this trial reported that enzalutamide treatment is feasible and well-tolerated in this patient population. Finally, phase I/II clinical trials continue to inform drug development and clinical practice, including trials of newer generation antiandrogen agents in women with AR+ breast cancer. Additional studies are needed to better understand use of antiandrogen therapies for the treatment of women with AR+ breast cancers.
Conclusion
While the role of AR in prostate cancer is more completely understood, the importance of AR signaling in breast cancer is an area of increasing investigation. In order to understand the mechanism of AR signaling and to design proper therapies against AR in breast cancer, additional work needs to be done to elucidate the mechanism by which AR is activating its target genes and contributing to tumor growth and metastasis, as well as systemic and radiation therapy resistance. Advancements in this mechanistic understanding will shed light on potential combination therapies and will allow for more effective treatment for patients with AR+ breast cancers. Further, discerning the intricacies and crosstalk between AR and ER signaling may also provide advancements for treatment of AR+, ER+ breast cancers. These outcomes would be impactful not only for the advanced understanding of the role of AR, but also for new ways in which AR signaling can be inhibited to improve outcomes for women with AR+ breast cancer.
References
Schweizer, M. T. & Yu, E. Y. AR-signaling in human malignancies: prostate cancer and beyond. Cancers 9, 1–19 (2017).
Hollenberg, S. M. et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318, 635–641 (1985).
Sullivan, W. P. et al. Isolation of steroid receptor binding protein from chicken oviduct and production of monoclonal antibodies. Biochemistry 24, 4214–4222 (1985).
Lindquist, S. & Craig, E. A. The heat-shock proteins. Annu. Rev. Genet. 22, 631–677 (1988).
Jäättelä, M. Heat shock proteins as cellular lifeguards. Ann. Med. 31, 261–271 (1999).
Sarker, D., Reid, A. H. M., Yap, T. A. & Bono, J. S. de. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 15, 4799–4805 (2009).
Visakorpi, T. et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 9, 401–406 (1995).
Gottlieb, B., Beitel, L. K., Nadarajah, A., Paliouras, M. & Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 33, 887–894 (2012).
Jensen, E. V. & Jordan, V. C. The estrogen receptor: a model for molecular medicine. Clin. Cancer Res. 9, 1980–1989 (2003).
Jensen, E. V., Block, G. E., Smith, S., Kyser, K. & DeSombre, E. R. Estrogen receptors and breast cancer response to adrenalectomy. J. Natl Cancer Inst. Monogr. 34, 55–70 (1971).
McGuire, W. L., Carbone, P. P. & Vollmer, E. P. Estrogen receptors in human breast cancer. (Raven Press New York, 1975).
Lumachi, F., Brunello, A., Maruzzo, M., Basso, U. & Basso, S. M. M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 20, 596–604 (2013).
Goss, P. E. et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N. Engl. J. Med. 349, 1793–1802 (2003).
Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351, 1451–1467 (1998).
Heuson, J. C. et al. Androgen Dependence of Breast Cancers. Lancet 302, 203–204 (1973).
Augello, M. A., Hickey, T. E. & Knudsen, K. E. FOXA1: master of steroid receptor function in cancer. EMBO J. 30, 3885–3894 (2011).
Parolia, A. et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418 (2019).
Zhang, C. et al. Definition of a FoxA1 cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res. 71, 6738–6748 (2011).
Sahu, B. et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 30, 3962–3976 (2011).
Robinson, J. L. L. et al. Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene 33, 5666–5674 (2014).
Laganière, J. et al. Location analysis of estrogen receptor α target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc. Natl Acad. Sci. 102, 11651–11656 (2005).
Robinson, J. L. L. et al. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J. 30, 3019–3027 (2011).
Hurtado, A., Holmes, K. A., Ross-Innes, C. S., Schmidt, D. & Carroll, J. S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 43, 27–33 (2011).
Guiu, S. et al. Coexpression of androgen receptor and FOXA1 in nonmetastatic triple-negative breast cancer: ancillary study from PACS08 trial. Future Oncol. Lond. Engl. 11, 2283–2297 (2015).
Guiu, S. et al. Prognostic value of androgen receptor and FOXA1 co-expression in non-metastatic triple negative breast cancer and correlation with other biomarkers. Br. J. Cancer 119, 76–79 (2018).
Robinson, J. L. L., Holmes, K. A. & Carroll, J. S. FOXA1 mutations in hormone-dependent cancers. Front. Oncol. 3, 1–6 (2013).
Wang, Y. et al. Differential regulation of PTEN expression by androgen receptor in prostate and breast cancers. Oncogene 30, 4327–4338 (2011).
Carver, B. S. et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19, 575–586 (2011).
Lin, H.-K., Hu, Y.-C., Lee, D. K. & Chang, C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol. Endocrinol. 18, 2409–2423 (2004).
El Sheikh, S. S., Romanska, H. M., Abel, P., Domin, J. & Lalani, E.-N. Predictive value of PTEN and AR coexpression of sustained responsiveness to hormonal therapy in prostate cancer—a pilot study. Neoplasia N. Y. N. 10, 949–953 (2008).
Wu, Z., Conaway, M., Gioeli, D., Weber, M. J. & Theodorescu, D. Conditional expression of PTEN alters the androgen responsiveness of prostate cancer cells. Prostate 66, 1114–1123 (2006).
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015).
Rizza, P. et al. Estrogen receptor beta as a novel target of androgen receptor action in breast cancer cell lines. Breast Cancer Res. BCR 16, R21 (2014).
Karamouzis, M. V., Papavassiliou, K. A., Adamopoulos, C. & Papavassiliou, A. G. Targeting androgen/estrogen receptors crosstalk in cancer. Trends Cancer 2, 35–48 (2016).
Whang, Y. E. et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl Acad. Sci. 95, 5246–5250 (1998).
Bedolla, R. et al. Determining risk of biochemical recurrence in prostate cancer by immunohistochemical detection of PTEN expression and Akt activation. Clin. Cancer Res. 13, 3860–3867 (2007).
Perren, A. et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am. J. Pathol. 155, 1253–1260 (1999).
Migliaccio, A. et al. Steroid-induced androgen receptor–oestradiol receptor β–Src complex triggers prostate cancer cell proliferation. EMBO J. 19, 5406–5417 (2000).
Leung, J. K. & Sadar, M. D. Non-genomic actions of the androgen receptor in prostate cancer. Front. Endocrinol. 8, 1–8 (2017).
Peterziel, H. et al. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene 18, 6322–6329 (1999).
Liao, R. S. et al. Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Transl. Androl. Urol. 2, 187–196 (2013).
Heinlein, C. A. & Chang, C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol. Endocrinol. 16, 2181–2187 (2002).
Thomas, P., Pang, Y., Dong, J. & Berg, A. H. Identification and characterization of membrane androgen receptors in the ZIP9 zinc transporter subfamily: II. Role of human ZIP9 in testosterone-induced prostate and breast cancer cell apoptosis. Endocrinology 155, 4250–4265 (2014).
Pi, M., Parrill, A. L. & Quarles, L. D. GPRC6A mediates the non-genomic effects of steroids. J. Biol. Chem. 285, 39953–39964 (2010).
Kalyvianaki, K. et al. Antagonizing effects of membrane-acting androgens on the eicosanoid receptor OXER1 in prostate cancer. Sci. Rep. 7, 44418 (2017).
Chia, K. M., Liu, J., Francis, G. D. & Naderi, A. A Feedback Loop between androgen receptor and ERK signaling in estrogen receptor-negative breast cancer. Neoplasia N. Y. N. 13, 154–166 (2011).
Bleach, R. & McIlroy, M. The divergent function of androgen receptor in breast cancer; analysis of steroid mediators and tumor intracrinology. Front. Endocrinol. 9, 1–19 (2018).
Khatun, A. et al. Transcriptional repression and protein degradation of the Ca2+-activated K+ channel KCa1.1 by androgen receptor inhibition in human breast cancer cells. Front. Physiol. 9, 1–12 (2018).
Ren, Q. et al. Expression of androgen receptor and its phosphorylated forms in breast cancer progression. Cancer 119, 2532–2540 (2013).
Roseweir, A. K. et al. Phosphorylation of androgen receptors at serine 515 is a potential prognostic marker for triple negative breast cancer. Oncotarget 8, 37172–37185 (2017).
Kampa, M., Pelekanou, V. & Castanas, E. Membrane-initiated steroid action in breast and prostate cancer. Steroids 73, 953–960 (2008).
Kalyvianaki, K. et al. Membrane androgen receptors (OXER1, GPRC6A AND ZIP9) in prostate and breast cancer: A comparative study of their expression. Steroids 142, 100–108 (2019).
Castoria, G. et al. Targeting androgen receptor/Src complex impairs the aggressive phenotype of human fibrosarcoma cells. PLoS ONE 8, 1–12 (2013).
Tilley, W. D., Marcelli, M., Wilson, J. D. & McPhaul, M. J. Characterization and expression of a cDNA encoding the human androgen receptor. Proc. Natl Acad. Sci. USA. 86, 327–331 (1989).
Hu, R. et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 69, 16–22 (2009).
Dehm, S. M., Schmidt, L. J., Heemers, H. V., Vessella, R. L. & Tindall, D. J. Splicing of a novel AR exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477 (2008).
Antonarakis, E., Armstrong, A., Dehm, S. & Luo, J. Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 19, 231–241 (2016).
Wadosky, K. M. & Koochekpour, S. Androgen receptor splice variants and prostate cancer: from bench to bedside. Oncotarget 8, 18550–18576 (2017).
Watson, P. A. et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl Acad. Sci. USA. 107, 16759–16765 (2010).
Hickey, T. E. et al. Expression of androgen receptor splice variants in clinical breast cancers. Oncotarget 6, 44728–44744 (2015).
Antonarakis, E. S. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 371, 1028–1038 (2014).
Szmulewitz, R. Z. et al. Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate 72, 157–164 (2012).
Arora, V. K. et al. Glucocorticoid receptor confers resistance to anti-androgens by bypassing androgen receptor blockade. Cell 155, 1309–1322 (2013).
Christenson, J. L. et al. Harnessing a different dependency: how to identify and target androgen receptor-positive versus quadruple-negative breast cancer. Horm. Cancer 9, 82–94 (2018).
Hu, D. G. et al. Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm. Cancer 5, 61–71 (2014).
Moore, N. L. et al. An androgen receptor mutation in the MDA-MB-453 cell line model of molecular apocrine breast cancer compromises receptor activity. Endocr. Relat. Cancer 19, 599–613 (2012).
Vera-Badillo, F. E. et al. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J. Natl Cancer Inst. 106, djt319 (2014).
Ogawa, Y. et al. Androgen receptor expression in breast cancer: relationship with clinicopathological factors and biomarkers. Int. J. Clin. Oncol. 13, 431–435 (2008).
Rangel, N. et al. The role of the AR/ER ratio in ER-positive breast cancer patients. Endocr. Relat. Cancer 25, 163–172 (2018).
Hammond, M. E. H. et al. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 28, 2784–2795 (2010).
Peters, A. A. et al. Androgen receptor inhibits estrogen receptor-α activity and is prognostic in breast cancer. Cancer Res. 69, 6131–6140 (2009).
Ricciardelli, C. et al. The magnitude of androgen receptor positivity in breast cancer is critical for reliable prediction of disease outcome. Clin. Cancer Res. 24, 2328–2341 (2018).
Hall, J. M. & McDonnell, D. P. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140, 5566–5578 (1999).
Cheng, J., Lee, E. J., Madison, L. D. & Lazennec, G. Expression of estrogen receptor β in prostate carcinoma cells inhibits invasion and proliferation and triggers apoptosis. FEBS Lett. 566, 169–172 (2004).
Hurtado, A. et al. Estrogen receptor beta displays cell cycle-dependent expression and regulates the G1 phase through a non-genomic mechanism in prostate carcinoma cells. Anal. Cell. Pathol. 30, 349–365 (2008).
Nakamura, Y. et al. Cyclin D1 (CCND1) expression is involved in estrogen receptor beta (ERβ) in human prostate cancer. Prostate 73, 590–595 (2013).
Christoforou, P., Christopoulos, P. F. & Koutsilieris, M. The role of estrogen receptor β in prostate cancer. Mol. Med. 20, 427–434 (2014).
De Amicis, F. et al. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 121, 1–11 (2010).
Cao, L. et al. A high AR:ERα or PDEF:ERα ratio predicts a sub-optimal response to tamoxifen therapy in ERα-positive breast cancer. Cancer Chemother. Pharmacol. 84, 609–620 (2019).
Chia, K. et al. Non-canonical AR activity facilitates endocrine resistance in breast cancer. Endocr. Relat. Cancer 26, 251–264 (2019).
D’Amato, N. C. et al. Cooperative dynamics of AR and ER activity in breast cancer. Mol. Cancer Res. 14, 1054–1067 (2016).
Cochrane, D. R. et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 16, R7 (2014).
Fioretti, F. M., Sita-Lumsden, A., Bevan, C. L. & Brooke, G. N. Revising the role of the androgen receptor in breast cancer. J. Mol. Endocrinol. 52, R257–R265 (2014).
Wilson, S., Qi, J. & Filipp, F. V. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep. 6, 32611 (2016).
Collins, L. C. et al. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod. Pathol. 24, 924–931 (2011).
Lehmann, B. D. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 121, 2750–2767 (2011).
Farmer, P. et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 24, 4660–4671 (2005).
Birrell, S. N. et al. Androgens induce divergent proliferative responses in human breast cancer cell lines. J. Steroid Biochem. Mol. Biol. 52, 459–467 (1995).
Ni, M. et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 20, 119–131 (2011).
Barton, V. N. et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol. Cancer Ther. 14, 769–778 (2015).
Wang, J. et al. ERβ1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res. Treat. 152, 255–269 (2015).
Anestis, A. et al. Estrogen receptor beta increases sensitivity to enzalutamide in androgen receptor-positive triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 145, 1221–1233 (2019).
Hall, R. E., Birrell, S. N., Tilley, W. D. & Sutherland, R. L. MDA-MB-453, an androgen-responsive human breast carcinoma cell line with high level androgen receptor expression. Eur. J. Cancer 30, 484–490 (1994).
Barton, V. N. et al. Androgen receptor supports an anchorage-independent, cancer stem cell-like population in triple-negative breast cancer. Cancer Res. 77, 3455–3466 (2017).
Naderi, A. & Hughes-Davies, L. A functionally significant cross-talk between androgen receptor and ErbB2 pathways in estrogen receptor negative breast cancer. Neoplasia N. Y. N. 10, 542–548 (2008).
Goodwin, J. F. et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 3, 1254–1271 (2013).
Polkinghorn, W. R. et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 3, 1245–1253 (2013).
Goodwin, J. F. et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell 28, 97–113 (2015).
Al-Ubaidi, F. L. T. et al. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 19, 1547–1556 (2013).
Speers, C. et al. Androgen receptor as a mediator and biomarker of radioresistance in triple-negative breast cancer. Npj Breast Cancer 3, 29 (2017).
Michmerhuizen, A. R. et al. Seviteronel, a Novel CYP17 lyase inhibitor and androgen receptor antagonist, radiosensitizes AR-positive triple negative breast cancer cells. Front. Endocrinol. 11, 1–13 (2020).
Yard, B. D. et al. A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nat. Commun. 7, 11428 (2016).
Sherr, C. J. & Roberts, J. M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18, 2699–2711 (2004).
Koryakina, Y., Knudsen, K. E. & Gioeli, D. Cell-cycle-dependent regulation of androgen receptor function. Endocr. Relat. Cancer 22, 249–264 (2015).
Balk, S. P. & Knudsen, K. E. AR, the cell cycle, and prostate cancer. Nucl. Recept. Signal. 6, 1–12 (2008).
Knudsen, K. E., Arden, K. C. & Cavenee, W. K. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J. Biol. Chem. 273, 20213–20222 (1998).
Xu, Y., Chen, S.-Y., Ross, K. N. & Balk, S. P. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 66, 7783–7792 (2006).
Yeh, S. et al. Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells. Biochem. Biophys. Res. Commun. 248, 361–367 (1998).
Lu, J. & Danielsen, M. Differential regulation of androgen and glucocorticoid receptors by retinoblastoma protein. J. Biol. Chem. 273, 31528–31533 (1998).
Lanzino, M. et al. Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells—identification of a novel androgen response element. Nucleic Acids Res. 38, 5351–5365 (2010).
Kokontis, J. M., Hay, N. & Liao, S. Progression of LNCaP prostate tumor cells during androgen deprivation: hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol. 12, 941–953 (1998).
Lu, S., Liu, M., Epner, D. E., Tsai, S. Y. & Tsai, M.-J. Androgen regulation of the cyclin-dependent kinase inhibitor p21 gene through an androgen response element in the proximal promoter. Mol. Endocrinol. 13, 376–384 (1999).
Wolf, D. A., Kohlhuber, F., Schulz, P., Fittler, F. & Eick, D. Transcriptional down-regulation of c-myc in human prostate carcinoma cells by the synthetic androgen mibolerone. Br. J. Cancer 65, 376–382 (1992).
Grad, J. M., Le Dai, J., Wu, S. & Burnstein, K. L. Multiple androgen response elements and a Myc consensus site in the androgen receptor (AR) coding region are involved in androgen-mediated up-regulation of AR messenger RNA. Mol. Endocrinol. 13, 1896–1911 (1999).
Zhu, A. et al. Antiproliferative effect of androgen receptor inhibition in mesenchymal stem-like triple-negative breast cancer. Cell. Physiol. Biochem. 38, 1003–1014 (2016).
Bièche, I., Parfait, B., Tozlu, S., Lidereau, R. & Vidaud, M. Quantitation of androgen receptor gene expression in sporadic breast tumors by real-time RT–PCR: evidence that MYC is an AR-regulated gene. Carcinogenesis 22, 1521–1526 (2001).
Bretones, G., Delgado, M. D. & León, J. Myc and cell cycle control. Biochim. Biophys. Acta. 1849, 506–516 (2015).
Amicis, F. D. et al. AIB1 sequestration by androgen receptor inhibits estrogen-dependent cyclin D1 expression in breast cancer cells. BMC Cancer 19, 1–11 (2019).
Anzick, S. L. et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277, 965–968 (1997).
List, H.-J., Reiter, R., Singh, B., Wellstein, A. & Riegel, A. T. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res. Treat. 68, 21–28 (2001).
Narbe, U. et al. The estrogen receptor coactivator AIB1 is a new putative prognostic biomarker in ER-positive/HER2-negative invasive lobular carcinoma of the breast. Breast Cancer Res. Treat. 175, 305–316 (2019).
Augello, M. A., Den, R. B. & Knudsen, K. E. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 33, 399–411 (2014).
Lin, C.-Y. et al. Elevation of androgen receptor promotes prostate cancer metastasis via induction of EMT and reduction of KAT5. Cancer Sci. 3564–3574 (2018).
Barron, D. A. & Rowley, D. R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 19, R187–R204 (2012).
Chan, J. S. K. et al. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 37, 160–173 (2018).
Yu, S. et al. Androgen receptor in human prostate cancer-associated fibroblasts promotes prostate cancer epithelial cell growth and invasion. Med. Oncol. 30, 674 (2013).
Ricke, E. A. et al. Androgen hormone action in prostatic carcinogenesis: stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 33, 1391–1398 (2012).
Leach, D. A. et al. Cell-lineage specificity and role of AP-1 in the prostate fibroblast androgen receptor cistrome. Mol. Cell. Endocrinol. 439, 261–272 (2017).
Chen, W.-Y. et al. Inhibition of the androgen receptor induces a novel tumor promoter, ZBTB46, for prostate cancer metastasis. Oncogene 36, 6213–6224 (2017).
Chen, J. et al. Androgen-deprivation therapy with enzalutamide enhances prostate cancer metastasis via decreasing the EPHB6 suppressor expression. Cancer Lett. 408, 155–163 (2017).
Giovannelli, P., Donato, M. D., Auricchio, F., Castoria, G. & Migliaccio, A. Androgens induce invasiveness of triple negative breast cancer cells through AR/Src/PI3-K complex assembly. Sci. Rep. 9, 1–14 (2019).
Feng, J. et al. Androgen and AR contribute to breast cancer development and metastasis: an insight of mechanisms. Oncogene 36, 2775–2790 (2017).
Grogg, A. et al. Androgen receptor status is highly conserved during tumor progression of breast cancer. BMC Cancer 15, 1–7 (2015).
Gasparini, P. et al. Androgen receptor status is a prognostic marker in non-basal triple negative breast cancers and determines novel therapeutic options. PLoS ONE 9, e88525 (2014).
Kraby, M. R. et al. The prognostic value of androgen receptors in breast cancer subtypes. Breast Cancer Res. Treat. 172, 283–296 (2018).
Bronte, G. et al. Androgen receptor expression in breast cancer: what differences between primary tumor and metastases? Transl. Oncol. 11, 950–956 (2018).
Perlmutter, M. A. & Lepor, H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev. Urol. 9, S3–S8 (2007).
Huggins, C., Stevens, R. E. & Hodges, C. V. Studies on prostate cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 43, 209–223 (1941).
Wilson, E. M. & French, F. S. Binding properties of androgen receptors. Evidence for identical receptors in rat testis, epididymis, and prostate. J. Biol. Chem. 251, 5620–5629 (1976).
Askew, E. B., Gampe, R. T., Stanley, T. B., Faggart, J. L. & Wilson, E. M. Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J. Biol. Chem. 282, 25801–25816 (2007).
Andriole, G. L. et al. Effect of the dual 5alpha-reductase inhibitor dutasteride on markers of tumor regression in prostate cancer. J. Urol. 172, 915–919 (2004).
Clark, R. V. et al. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J. Clin. Endocrinol. Metab. 89, 2179–2184 (2004).
McConnell, J. D. et al. Finasteride, an inhibitor of 5 alpha-reductase, suppresses prostatic dihydrotestosterone in men with benign prostatic hyperplasia. J. Clin. Endocrinol. Metab. 74, 505–508 (1992).
Span, P. N. et al. Selectivity of finasteride as an in vivo inhibitor of 5alpha-reductase isozyme enzymatic activity in the human prostate. J. Urol. 161, 332–337 (1999).
Wu, Y. et al. Prostate cancer cells differ in testosterone accumulation, dihydrotestosterone conversion, and androgen receptor signaling response to steroid 5α-reductase inhibitors. Prostate 73, 1470–1482 (2013).
Mina, A., Yoder, R. & Sharma, P. Targeting the androgen receptor in triple-negative breast cancer: current perspectives. OncoTargets Ther. 10, 4675–4685 (2017).
Ryan, C. J. et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 16, 152–160 (2015).
Bonnefoi, H. et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann. Oncol. 27, 812–818 (2016).
Proverbs-Singh, T., Feldman, J. L., Morris, M. J., Autio, K. A. & Traina, T. A. Targeting the androgen receptor in prostate and breast cancer: several new agents in development. Endocr. Relat. Cancer 22, R87–R106 (2015).
Montgomery, R. B. et al. Phase I clinical trial of galeterone (TOK-001), a multifunctional antiandrogen and CYP17 inhibitor in castration resistant prostate cancer (CRPC). J. Clin. Oncol. 30, 4665–4665 (2012).
Hook, K. V., Huang, T. & Alumkal, J. J. Orteronel for the treatment of prostate cancer. Future Oncol. 10, 803–811 (2014).
Fizazi, K. et al. Phase III, randomized, double-blind, multicenter trial comparing orteronel (TAK-700) plus prednisone with placebo plus prednisone in patients with metastatic castration-resistant prostate cancer that has progressed during or after docetaxel-based therapy: ELM-PC 5. J. Clin. Oncol. 33, 723–731 (2015).
Rampurwala, M. M. et al. Phase 1b study of orteronel in postmenopausal women with hormone-receptor positive (HR+) metastatic breast cancer. J. Clin. Oncol. 32, 538–538 (2014).
Takeda Announces Termination of Orteronel (TAK-700) Development for Prostate Cancer in Japan, U.S.A. and Europe. Takeda https://www.takeda.com/newsroom/newsreleases/2014/takeda-announces-termination-of-orteronel-tak-700-development-for-prostate-cancer-in-japan-u.s.a.-and-europe/ (2014).
Gucalp, A. & Traina, T. A. Targeting the androgen receptor in triple-negative breast cancer. Curr. Probl. Cancer 40, 141–150 (2016).
Perrault, D. J. et al. Phase II study of flutamide in patients with metastatic breast cancer. A National Cancer Institute of Canada Clinical Trials Group study. Invest. N. Drugs 6, 207–210 (1988).
Barqawi, A., Akduman, B., Abouelfadel, Z., Robischon, M. & Crawford, E. D. The use of flutamide as a single antiandrogen treatment for hormone-refractory prostate cancer. BJU Int. 92, 695–698 (2003).
Gucalp, A. et al. Phase II trial of bicalutamide in patients with androgen receptor–positive, estrogen receptor–negative metastatic breast cancer. Clin. Cancer Res. 19, 5505–5512 (2013).
Veldscholte, J. et al. A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 173, 534–540 (1990).
Yeh, S., Miyamoto, H. & Chang, C. Hydroxyflutamide may not always be a pure antiandrogen. Lancet 349, 852–853 (1997).
Lu, Q. et al. Bicalutamide plus aromatase inhibitor in patients with estrogen receptor‐positive/androgen receptor‐positive advanced breast cancer. Oncologist 21, 21–e15 (2020).
Caiazza, F. et al. Preclinical evaluation of the AR inhibitor enzalutamide in triple-negative breast cancer cells. Endocr. Relat. Cancer 23, 323–334 (2016).
Scher, H. I. et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N Engl J Med. 367, 1187–1197 (2012).
Traina, T. A. et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J. Clin. Oncol. 33, 2141–2147 (2015).
Chong, J. T., Oh, W. K. & Liaw, B. C. Profile of apalutamide in the treatment of metastatic castration-resistant prostate cancer: evidence to date. OncoTargets Therapy https://www.dovepress.com/profile-of-apalutamide-in-the-treatment-of-metastatic-castration-resis-peer-reviewed-article-OTT (2018). https://doi.org/10.2147/OTT.S147168.
Clegg, N. J. et al. ARN-509: a novel anti-androgen for prostate cancer treatment. Cancer Res. 72, 1494–1503 (2012).
Smith, M. R. et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N. Engl. J. Med. 378, 1408–1418 (2018).
Moilanen, A.-M. et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 5, 12007 (2015).
Fizazi, K. et al. Darolutamide in nonmetastatic, castration-resistant. Prostate Cancer N. Engl. J. Med. 380, 1235–1246 (2019).
Christenson, J. L. et al. Harnessing a different dependency: how to identify and target androgen receptor-positive versus quadruple-negative breast cancer. Horm. Cancer 9, 82–94 (2018).
Bardia, A. et al. Phase 1 study of seviteronel, a selective CYP17 lyase and androgen receptor inhibitor, in women with estrogen receptor-positive or triple-negative breast cancer. Breast Cancer Res. Treat. 1–10. https://doi.org/10.1007/s10549-018-4813-z (2018).
Gupta, S. et al. Phase 1 study of seviteronel, a selective CYP17 lyase and androgen receptor inhibitor, in men with castration-resistant prostate cancer. Clin. Cancer Res. 0564.2018. https://doi.org/10.1158/1078-0432.CCR-18-0564 (2018).
Reese, J. M. et al. Abstract P5-05-05: Targeting the androgen receptor with seviteronel, a CYP17 lyase and AR inhibitor, in triple negative breast cancer. Cancer Res. 79. https://doi.org/10.1158/1538-7445.SABCS18-P5-05-05 (2019).
Gucalp, A. et al. Phase (Ph) 2 stage 1 clinical activity of seviteronel, a selective CYP17-lyase and androgen receptor (AR) inhibitor, in women with advanced AR+ triple-negative breast cancer (TNBC) or estrogen receptor (ER)+ BC: CLARITY-01. J. Clin. Oncol. 35, 1102–1102 (2017).
Gucalp, A. et al. Abstract P2-08-04: Phase 1/2 study of oral seviteronel (VT-464), a dual CYP17-lyase inhibitor and androgen receptor (AR) antagonist, in patients with advanced AR positive triple negative (TNBC) or estrogen receptor (ER) positive breast cancer (BC). Cancer Res. 77. https://doi.org/10.1158/1538-7445.SABCS16-P2-08-04 (2017).
Zweifel, M. et al. Phase I trial of the androgen receptor modulator CR1447 in breast cancer patients. Endocr. Connect. 6, 549–556 (2017).
Srinath, R. & Dobs, A. Enobosarm (GTx-024, S-22): a potential treatment for cachexia. Future Oncol. 10, 187–194 (2014).
Dalton, J. T. et al. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J. Cachexia Sarcopenia Muscle 2, 153–161 (2011).
Dobs, A. S. et al. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol. 14, 335–345 (2013).
Overmoyer, B. et al. Enobosarm: A targeted therapy for metastatic, androgen receptor positive, breast cancer. J. Clin. Oncol. 32, 568–568 (2014).
Goldenberg, I. S. Testosterone propionate therapy in breast cancer. JAMA 188, 1069–1072 (1964).
Lim, E. et al. Pushing estrogen receptor around in breast cancer. Endocr. Relat. Cancer 23, T227–T241 (2016).
Lee-Bitar, J. S. et al. A phase II clinical trial of pembrolizumab and selective androgen receptor modulator GTx-024 in patients with advanced androgen receptor-positive triple-negative breast cancer. J. Clin. Oncol. 37, 1069–1069 (2019).
Eder, I. E. et al. Inhibition of LNCaP prostate cancer cells by means of androgen receptor antisense oligonucleotides. Cancer Gene Ther. 7, 997–1007 (2000).
Eder, I. E. et al. Inhibition of LNCaP prostate tumor growth in vivo by an antisense oligonucleotide directed against the human androgen receptor. Cancer Gene Ther. 9, 117–125 (2002).
Hamy, F. et al. Specific block of androgen receptor activity by antisense oligonucleotides. Prostate Cancer Prostatic Dis. 6, 27–33 (2003).
Velez, M. V. L., Verhaegh, G. W., Smit, F., Sedelaar, J. P. M. & Schalken, J. A. Suppression of prostate tumor cell survival by antisense oligonucleotide-mediated inhibition of AR-V7 mRNA synthesis. Oncogene 1. https://doi.org/10.1038/s41388-019-0696-7 (2019).
Zou, Y., Ma, D. & Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 37, 21–30 (2019).
Han, X. et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 62, 941–964 (2019).
Han, X. et al. Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. J. Med. Chem. 62, 11218–11231 (2019).
Kregel, S. et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia N. Y. N. 22, 111–119 (2020).
Salami, J. et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 1, 1–9 (2018).
Eil, C. Ketoconazole binds to the human androgen receptor. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 24, 367–370 (1992).
Zhang, S. et al. Endocrine disruptors of inhibiting testicular 3β-hydroxysteroid dehydrogenase. Chem. Biol. Interact. 303, 90–97 (2019).
Lim, Y.-W. et al. Pharmacokinetics and pharmacodynamics of docetaxel with or without ketoconazole modulation in chemonaive breast cancer patients. Ann. Oncol. 21, 2175–2182 (2010).
Rathkopf, D. E. et al. An open label phase 1/2A study to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy of TRC253, an androgen receptor antagonist, in patients with metastatic castration-resistant prostate cancer. JCO. 37, e16542–e16542 (2019).
Joseph, J. D. et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 3, 1020–1029 (2013).
Spratt, D. E. et al. Androgen receptor upregulation mediates radioresistance after ionizing radiation. Cancer Res. 75, 4688–4696 (2015).
Ghashghaei, M. et al. Enhanced radiosensitization of enzalutamide via schedule dependent administration to androgen-sensitive prostate cancer cells. Prostate 78, 64–75 (2018).
Manem, V. S. et al. Modeling cellular response in large-scale radiogenomic databases to advance precision radiotherapy. Cancer Res. 79, 6227–6237 (2019).
Schiewer, M. J. et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2, 1134–1149 (2012).
Li, L. et al. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal 10, eaam7479 (2017).
Asim, M. et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 8, 374 (2017).
Pamarthy, S. et al. Abstract 1114: combining anti-androgen therapy and PARP inhibition results in synergistic cytotoxicity of metastatic castration-resistant prostate cancer (mCRPC) cells. Cancer Res. 77, 1114–1114 (2017).
Karanika, S., Karantanos, T., Li, L., Corn, P. G. & Thompson, T. C. DNA damage response and prostate cancer: defects, regulation and therapeutic implications. Oncogene 34, 2815–2822 (2015).
Comen, E. & Robson, M. Poly(ADP-Ribose) polymerase inhibitors in triple-negative breast cancer. Cancer J. 16, 48–52 (2010).
Murai, J. et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599 (2012).
Comstock, C. E. S. et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene 32, 5481–5491 (2013).
Gucalp, A. et al. Abstract P3-11-04: Phase I/II trial of palbociclib in combination with bicalutamide for the treatment of androgen receptor (AR)+ metastatic breast cancer (MBC). Cancer Res. 78. https://doi.org/10.1158/1538-7445.SABCS17-P3-11-04 (2018).
Qi, W. et al. Reciprocal feedback inhibition of the androgen receptor and PI3K as a novel therapy for castrate-sensitive and -resistant prostate cancer. Oncotarget 6, 41976–41987 (2015).
Lehmann, B. D. et al. TBCRC 032 IB/II multicenter study: molecular insights to AR antagonist and PI3K inhibitor efficacy in patients with AR+ metastatic triple-negative breast cancer. Clin. Cancer Res. 26, 2111–2123 (2020).
Traina, T. A. et al. Adjuvant enzalutamide for the treatment of early-stage androgen receptor-positive (AR+) TNBC. J. Clin. Oncol. 37, 546–546 (2019).
Traina, T. A. et al. Abstract P5-12-09: Patient-reported outcomes (PROs) during one year of adjuvant enzalutamide for the treatment of early stage androgen receptor positive (AR+) triple negative breast cancer. Cancer Res. 80. https://doi.org/10.1158/1538-7445.SABCS19-P5-12-09 (2020).
Acknowledgements
The authors thank Steve Kronenberg for his assistance in creating the figure and illustration. A.R.M. is supported by T32-GM007315. Funding for this work was generously provided in part by a grant from the Breast Cancer Research Foundation (N02600 to L.J.P.) and the University of Michigan Rogel Cancer Center Breast Strategic Fund (P30 F049977 to C.S.).
Author information
Authors and Affiliations
Contributions
A.R.M., D.E.S., L.J.P., C.S. all contributed to the concept inception, review of literature, writing, editing, and approval of the final version.
Data sharing
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Michmerhuizen, A.R., Spratt, D.E., Pierce, L.J. et al. ARe we there yet? Understanding androgen receptor signaling in breast cancer. npj Breast Cancer 6, 47 (2020). https://doi.org/10.1038/s41523-020-00190-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-020-00190-9
This article is cited by
-
The scaffold protein disabled 2 (DAB2) and its role in tumor development and progression
Molecular Biology Reports (2024)
-
Co-expression of Twist and Snai1: predictor of poor prognosis and biomarker of treatment resistance in untreated prostate cancer
Molecular Biology Reports (2024)
-
Imagine beyond: recent breakthroughs and next challenges in mammary gland biology and breast cancer research
Journal of Mammary Gland Biology and Neoplasia (2023)
-
Correlation between androgen receptor expression and pathological response rate in pre-operative HER2-positive breast cancer patients
Journal of Cancer Research and Clinical Oncology (2023)
-
Androgens exacerbate hepatic triglyceride accumulation in rats with polycystic ovary syndrome by downregulating MTTP expression
Endocrine (2023)
