
Abstract
Most prostate cancers express the androgen receptor (AR), and tumor growth and progression are facilitated by exceptionally low levels of systemic or intratumorally produced androgens. Thus, absolute inhibition of the androgen signaling axis remains the goal of current therapeutic approaches to treat prostate cancer (PCa). Paradoxically, high dose androgens also exhibit considerable efficacy as a treatment modality in patients with late-stage metastatic PCa. Here we show that low levels of androgens, functioning through an AR monomer, facilitate a non-genomic activation of the mTOR signaling pathway to drive proliferation. Conversely, high dose androgens facilitate the formation of AR dimers/oligomers to suppress c-MYC expression, inhibit proliferation and drive a transcriptional program associated with a differentiated phenotype. These findings highlight the inherent liabilities in current approaches used to inhibit AR action in PCa and are instructive as to strategies that can be used to develop new therapeutics for this disease and other androgenopathies.
Similar content being viewed by others
Introduction
Androgens, acting through the androgen receptor (AR), are involved in the development of the male reproductive system, manifestation of secondary sex characteristics at puberty, and establishment and maintenance of reproductive function1. They are also key regulators of metabolic homeostasis and processes required for normal skeletal/bone integrity and muscle development/function2,3,4,5,6. Dysregulated responses to AR/androgens are a pathogenomic feature of prostate cancer (PCa)7. The primary androgen, testosterone (T), is synthesized de novo in the Leydig cells within the testes and can be transformed to the higher affinity androgen dihydrotestosterone (DHT) by any of three genetically distinct 5α-reductases (SRD5A1-3)8,9. A long-standing question in developmental biology is how and why some phenotypic responses in males require only low levels of T whereas others more associated with differentiated phenotypes require higher levels of this hormone or exposure to DHT10. Similarly, the response of prostate tumors to androgens also differs as a function of dose11,12. These observations suggest that AR-expressing cells must possess mechanisms that enable them to recognize and respond differently to different levels of androgenic ligands13,14.
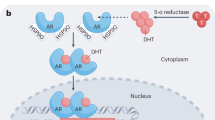
In contemporary models of AR action/pharmacology it is posited that in the absence of ligand, AR resides in the cytoplasm in an inactive form bound to a large multi-factor heat-shock protein (HSP) complex15. Upon binding an agonist, the receptor undergoes a conformational change that results in its displacement from HSPs and subsequent dimerization. The dimeric receptor then enters the nucleus, interacts with specific androgen response elements (AREs) located within the regulatory regions of target genes, and recruits functionally distinct coregulators to positively or negatively regulate target gene transcription16. Whereas this model is similar to that described for other nuclear receptors (NRs), there are important differences in how AR interacts with its attendant coregulators. For most NRs (a) agonist binding enables the formation of the primary coregulator binding pocket (activation function-2; AF-2) in the ligand binding domain of the receptor, enabling it to interact with coregulators that contain specific LXXLL motif(s)17,18,19,20 and (b) subtle differences in the structure of bound ligands can alter the shape of the AF-2 pocket allowing differential engagement of coregulators21,22. AR does not contain a classical AF-2 pocket, but rather agonist binding results in the formation of a shallow hydrophobic pocket (in a similar region to the AF-2 pocket of other NRs) that enables an intramolecular interaction with an FXXLF motif at the amino terminus of the receptor or with cofactors that contain an FXXLF motif23,24,25,26,27,28,29,30. A recently completed cryoEM structure of an AR agonist-coregulator complex on DNA revealed that two monomers of AR (each forming an intramolecular N/C terminal interaction) dimerize forming an asymmetric coregulator binding surface that accommodates one molecule each of a p160 coregulator (i.e., SRC-3) and p30031. In this dimeric complex, the FXXLF binding domain within AR is engaged in the formation of an N/C terminal interaction and is not available to interact with FXXLF motif containing proteins24. Within the confines of this inferred mechanism of action, it is unclear how AR actions can be supported by extremely low levels of systemic or intratumoral androgens in patients with PCa and conversely how treatment with high-dose androgens is an effective strategy in patients with late-stage disease32,33,34,35,36,37. Defining the molecular basis for this paradoxical pharmacology was the primary objective of this study.
Using a combination of genetic, biochemical, and chemical approaches, we show here that different doses of the same androgen alter the relative abundance of AR monomers and dimers/oligomers in cells and that these different forms of the receptor participate in distinct signaling pathways to exhibit unique biological activities. Thus, the oligomerization state of AR constitutes a biosensor that enables cells to recognize and respond in a distinct manner to different levels of the same hormone.
Results
Proliferative responses to different doses of androgens occur in a non-linear manner
As a starting point, we revisited and confirmed the longstanding observation that low-dose (LD) androgens (modeling the hypogonadal state) stimulate and high-dose (HD) androgens (modeling the eugonadal state) inhibit the proliferation of AR-positive PCa cell lines in vitro when tested in media where serum androgens have been removed (Fig. 1a, b)38,39. In fetal bovine serum (FBS) containing media, which contains endogenous androgens and other growth factors, we determined that HD androgens effectively inhibit proliferation (Fig. 1c, d). The synthetic AR agonist R1881 was used for these initial studies, although T and DHT function similarly (Fig. S1A, B). Non-linear dose responses to T (Fig. 1e) or DHT (Fig. 1f) were also observed in vivo in the LNCaP and VCaP (Fig. S1C) xenograft models of PCa.

LNCaP (a) or VCaP (b) cells plated in media containing charcoal-stripped serum (CFS) or full-serum (FBS) (c, d) were treated with increasing concentrations of R1881. Cell growth was assessed by DNA quantification at day 7. Data are shown as mean ± SD as representative results from three independent experiments, n = 3 wells of cells. RFU relative fluorescence units. Castrated male NSG mice bearing subcutaneous LNCaP tumors were administered increasing doses of testosterone in (e) (Placebo, n = 10 mice; 0.5 mg/kg/d, n = 9 mice; 1 mg/kg/d, n = 10 mice; 2 mg/kg/d, n = 10 mice; 4 mg/kg/d, n = 9 mice; 8 mg/kg/d, n = 9 mice) or DHT in (f) Placebo, n = 10; 0.1 mg/kg/d, n = 10 mice; 0.2 mg/kg/d, n = 10 mice; 0.4 mg/kg/d, n = 10 mice; 0.8 mg/kg/d, n = 10 mice. Data are shown as mean ± SD. p values were determined by two-way ANOVA followed by Tukey’s multiple comparison test (P < 0.0001 for (e) and P < 0.0004 for (f)). g Significant Hallmark pathways of selected Gene Set Enrichment Analysis (GSEA) differentially regulated in VCaP cells in response to low (0.06 nM R1881) as compared to high dose (R1881 10 nM) androgens are represented (FDR q value < 0.001; NES normalized enrichment score). NES < 0 represents downregulation of specified pathway in LD vs HD. NES > 0 represents upregulation of specified pathway in LD vs HD. h VCaP cells plated in CFS-supplemented media were treated with increasing concentrations of R1881 for 24 h. The mRNA expression for PSA and E2F1 were assessed using qRT-PCR. Data are shown as mean ± SD as representative results from three independent experiments, n = 3 technical replicates. i Heatmap presentation of gene expression as assessed using the Nanostring nCounter platform from VCaP cells treated in CFS-supplemented media with either vehicle (Veh), LD or HD R1881. j VCaP cells plated in media supplemented with CFS were treated with increasing concentrations of either R1881, Testosterone (T) or Dihydrotestosterone (DHT) for 48 h. Whole cell extracts were probed for RB1 phosphorylation (pRB), E2F1, FOXM1, PSA or β-Actin using western blot. Representative images are shown from n = 3 biologically independent experiments. Source data are provided as a Source Data file.
To explore the molecular mechanisms underlying the biphasic response to androgens, we performed RNA-seq in both LNCaP and VCaP cells following treatment with vehicle or increasing concentrations of androgens (from LD to HD) for 24 h. This analysis revealed striking non-linear dose-dependent differences in gene expression profiles. Notable were a group of mRNAs that encode proteins involved in cell proliferation (E2F targets, G2M checkpoint e.g., E2F1) and c-MYC biology, all of which were induced in response to LD androgens and inhibited by HD androgens (Figs. 1g, h and S1, D, F and G). In contrast, the classical AR target genes (e.g., PSA) were strongly induced in a linear manner in response to HD androgens (Figs. 1g, h and S1E). Using the Nanostring nCounter platform, we evaluated the expression of 327 AR-regulated genes identified in the RNA-seq analysis and confirmed their biphasic gene expression pattern in response to androgen dose in VCaP cells (Figs. 1i, and S1H). Similar biphasic responses were noted in LNCaP cells (Fig. S2A). Importantly, androgen-induced expression of LD mRNAs also resulted in a substantial increase in the expression of their corresponding proteins (e.g., E2F1 and FOXM1), and this was coincident with increased phosphorylation of RB (Fig. 1j). The expression of PSA was only observed in HD androgen-treated cells, and under those conditions, a dramatic downregulation of E2F1 and FOXM1 expression and a complete loss of RB phosphorylation was observed (Fig. 1j). Importantly, intratumoral levels of PSA increase in a linear, dose proportional, manner to androgens whereas tumor growth in the same animals exhibits a biphasic response (Figs. S1C and S2B). Thus, AR-expressing PCa cells/tumors have an inherent ability to recognize and respond differently to different levels of androgens.
Androgen-dependent proliferation of PCa cells is uncoupled by dose from canonical AR transcriptional activity
To identify molecular events responsible for the non-linear proliferative responses to androgens, we performed an unbiased genome-wide survey of changes in chromatin accessibility with the goal of identifying potential cis-acting elements enriched in LD versus HD treated PCa cells. To this end, we performed an assay for transposase-accessible chromatin using sequencing (ATAC-seq) in both VCaP and LNCaP cells following treatment with vehicle, or increasing concentrations of androgens for 24 h. In this manner, a total of 7810 and 104,198 high-confidence differentially accessible peaks were identified in androgen versus vehicle-treated VCaP and LNCaP cells respectively (Fig. 2a–d). Surprisingly, no significant peaks (when compared to vehicle) were identified in the chromatin isolated from VCaP cells treated with lower doses of androgens (Fig. 2a, c), which achieved significant proliferative responses (0.01 and 0.03 nM) (Fig. 1b), although the RNA pathway for E2F targets is clearly upregulated at these doses (Figs. 1h, i, and S1D). Increased differentially accessible sites in VCaP cells were only present at a dose of 0.06 nM R1881 (535 up and 33 sites down) and all but 3 of these sites, were common to HD (10 nM R1881; 6293 up and 1514 down sites) (Fig. 2a, c). In LNCaP cells, changes in chromatin architecture could be detected at all R1881 doses tested (0.03, 0.06, 0.1, and 10 nM), however, most of the sites identified were common to HD (10 nM R1881: 103,408 sites; 73,415 up and 29,993 down) and the magnitude of the peaks followed a classical linear response relationship (Fig. 2b, d). Motif analysis of differentially accessible peaks observed only in HD (10 nM R1881) or common to both HD and LD treated VCaP (0.06 nM), or LNCaP (0.03, 0.06, 0.1 nM) cells (Fig. 2a–d) showed that, as expected, AREs, FOXA1 and HoxB13 binding sequences were among the top enriched motifs (Supplementary Data 1 and Supplementary Data 2). These results are consistent with previously published reports and serve as validation of the ATAC-seq data40. Specific to LNCaP cells, a subset of uniquely enriched peaks was observed in response to LD treatments (428 up and 361 down sites; Fig. 2b, d). These sequences were also found to be enriched for AREs, FOXA1, and HoxB13 motifs (Fig. 2b, d and Supplementary Data 1 and Supplementary Data 2), however, no biological pathways were associated with these sites. The chromatin architecture in genes that comprised the classic Hallmark Androgen Response revealed peaks the magnitude of which followed a classical linear response relationship (Fig. S3A). A more focused analysis of Hallmark E2F targets, (expressed in response to LD androgens (Fig. S1D)), importantly, failed to identify any significant differentially accessible chromatin sites specific to LD androgen (Fig. S3B). Further, as opposed to the classical AR-regulated genes (e.g., PSA, FKBP5, and STEAP4), analysis of the genomic tracks of select genes induced in response to LD androgens (i.e., Hallmark E2F targets E2F1; CDC6 and FOXM1), did not show any changes in chromatin accessibility when evaluated 50 kb upstream or downstream of the transcription start site (TSS) (Figs. 2e and S3C). Thus, we conclude that whereas AR is required for the proliferation of PCa cells in response to LD androgens, the results from our ATAC-seq analysis suggest that in the presence of LD androgen, AR is likely acting in a non-canonical manner to facilitate proliferative responses as opposed to the well-described classical model of HD androgen action.

Heatmap of ATAC-seq signal within a 6 kb window of all high-confidence differentially accessible peaks identified in response to indicated androgen dose compared to vehicle treated VCaP (a) or LNCaP (b) cells. Differentially accessible peaks were subdivided based on whether they are significantly increased (VCaP Up or LNCaP Up) or significantly decreased (VCaP down or LNCaP down). Quantification of unique and common high-confidence differentially accessible peaks identified in response to indicated androgen dose compared to vehicle treated VCaP (c) or LNCaP (d) cells. e Representative genomic tracks of ATAC-seq sites within classical AR regulated genes (i.e., KLK3, FKBP5 and STEAP4) or LD regulated genes (i.e., E2F1, CDC6 and FOXM1) in response to vehicle (0) or increasing androgens in VCaP cells.
Androgen-dependent proliferation of PCa cells and interaction of AR with chromatin are uncoupled by androgen dose
The observation that low-dose androgens did not induce any significant changes in chromatin accessibility (Fig. 2) was surprising given that at these concentrations upregulated E2F target gene expression and cell proliferation were apparent (Figs. 1b, h and i and S1D). Thus, a genome-wide ChIP-sequencing (ChIP-seq) study was conducted in both VCaP and LNCaP cells with the goal of assessing AR binding at the enhancers of genes encoding proteins that were responsible for the non-linear (proliferative and growth inhibitory) responses to androgens. In this manner, a total of 39,621 and 53,871 high-confidence differentially bound sites were identified in androgen versus vehicle-treated VCaP and LNCaP respectively (Fig. 3a–d). In concordance with the ATAC-seq data, no specific differentially bound sites were identified in VCaP cells treated with the lowest doses of androgens (Fig. 3a, c), even though these doses promoted a significant proliferative response (0.01 and 0.03 nM) (Fig. 1b) and upregulated the RNA pathway for E2F targets (Figs. 1h, i, and S1D). Sites at which AR was bound differentially in response to 0.06 nM R1881 (5245 up and 2 sites down) were all common to HD androgen (10 nM R1881; 37,591 up and 2025 down sites) in all but 4 sites (Fig. 3a, c). Similarly, in LNCaP cells, fewer AR binding peaks were detected in chromatin isolated from cells treated with lower doses of androgen (0.06 and 0.1 nM R1881; 13,843 up and 10 down); however, all but, 85 of the sites identified were common to HD hormone (10 nM R1881: 53,786 sites; 52,249 up and 1537 down) and the magnitude of the peaks followed a classical linear response relationship (Fig. 3b, d). Consistent with previously published reports, motif analysis of differentially AR bound peaks observed in response to HD (10 nM R1881), or common to both HD and LD treated VCAP or LNCAP cells, revealed AREs, FOXA1 and HoxB13 binding sequences as the top enriched motifs40. A focused analysis of the genomic tracks of select classical HD-regulated genes (e.g., PSA, FKBP5 and STEAP4) revealed peaks where the degree of AR binding followed a classical linear response relationship (Figs. 3e and S4). However, a similar analysis of the genomic tracks of select genes which we have shown to be induced in response to LD androgens (e.g., Hallmark E2F targets E2F1; CDC6 and FOXM1), did not show any changes in AR bound peaks sites specific to LD androgen when evaluated 50 kb upstream or downstream of the TSS (Figs. 3e and S4). Thus, the results from our ChIP-seq analysis suggest that the degree of AR-binding to chromatin does not correlate with the expression of genes required for proliferation but does correlate with the expression of more classically regulated AR target genes (e.g., PSA) in HD conditions.

Heatmap of ChIP-seq signal within a 6 kb window of all high-confidence differentially accessible peaks identified in response to high dose (HD) and low dose (LD) androgen compared to vehicle treated VCaP (a) or LNCaP (b) cells. Differentially accessible peaks were subdivided based on whether they are significantly increased (VCaP Up or LNCaP Up) or decreased (VCaP down or LNCaP down). Quantification of unique and common high-confidence differentially accessible peaks identified in response to androgen dose compared to vehicle treated VCaP (c) or LNCaP (d) cells. e Representative genomic tracks of AR binding sites within HD- (KLK3, FKBP5 and STEAP4) or LD- androgen regulated genes (i.e., E2F1, CDC6 and FOXM1) in response to vehicle (0) or increasing androgen concentrations in VCaP cells.
Androgen-dependent proliferation of PCa cells does not require AR dimerization
The results of our RNA-seq, ATAC-seq, and ChIP-seq analysis do not support the established canonical model of AR action, where it is posited that a high fractional saturation of the receptor is required to enable the formation of AR homodimers, nuclear translocation, coregulator recruitment and transcriptional regulation of target genes41. Thus, it was difficult to explain how biological responses to androgens could occur in LD conditions (low fractional saturation). At odds with this model of AR action, our data suggested that AR dimers may not in fact be required for cell proliferation. To probe this pharmacology further, we first confirmed that the proliferative response to LD/HD T and DHT in VCaP cells were similar to what we described for R1881 (Fig. 4a). Subsequently, a quantitative cell-based NanoBiT assay was developed and used to evaluate AR dimerization as a function of ligand exposure42. To this end SmBiT-AR and LgBiT-AR chimera were expressed in HEK293 cells enabling a quantitative, real-time, assessment of the effects of ligand and dose on receptor dimerization (Fig. 4b). Importantly, minimal, if any dimerization of AR was observed at doses below 1 nM for any of the androgens tested in this assay (Fig. 4c). Using the same assay, a similar dimerization pattern was observed in response to different doses of R1881 in LNCaP cells (Fig. S5A). Further, it was noted that AR does not form dimers in media supplemented with FBS unless HD androgens were added supporting the idea that FBS stimulated cell growth is driven by LD androgens and likely mediated by AR monomers (Fig. S5B). Collectively, these data indicate that the proliferative responses to LD androgens occur at doses of hormone that are below that needed for dimerization, implying that these responses are likely mediated by a monomeric form of AR.

a VCaP cells were plated in CFS-supplemented media for 2 days followed by treatment with increasing concentrations of R1881, DHT or Testosterone (T). Cell growth was assessed by DNA quantification at day 7. Data are shown as mean ± SD of representative results from three independent experiments, n = 3 wells of cells. RFU: relative fluorescence units. b Schematic of the NanoBiT-based assay used to evaluate wtAR dimerization. c HEK293 cells were transfected in CFS-supplemented media with plasmids expressing full-length AR fused to the small- (SmBiT AR) and large-bit (LgBiT AR) of Nanoluciferase. 48 h later, cells were treated with increasing concentrations of R1881, DHT or testosterone (T) and at the same time Nanoluciferase reagent was added and light emission measured using a plate reader. Data are plotted as mean ± SD of representative results from four independent experiments, n = 3 wells of cells. RLU relative light units. Source data are provided as a Source Data file.
Identification of AR ligands which induce proliferation but do not facilitate AR dimerization
To probe the role of dimerization in LD/HD androgen signaling we first recreated previously described AR mutants that have been reported to interfere with dimerization43. Unfortunately, all of the mutants we generated which compromised dimerization also exhibited substantial loss of ligand binding affinity/transcriptional activity and thus were not useful for our studies. Thus, we undertook an alternative chemical biology approach to address this mechanistic question. To this end, we used the NanoBiT AR dimerization assay to screen a small molecule library of previously characterized AR ligands14,19,44. We hypothesized that it would be possible to identify AR ligands that induce PCa cell proliferation without inducing AR dimerization. Using this approach, we identified RU486 (Fig. 5a), an antiprogestin/antiglucocorticoid that we and others have shown to bind AR and effectively inhibit androgen action at classical target genes but which has been shown to be ineffective as a PCa therapeutic19,45,46. The synthetic steroidal AR ligand RTI001 (among others) was also identified and unlike RU486 this drug does not interact with GR or ERβ (Fig. 5a)47. Follow-up studies using a NanoBiT assay confirmed that neither RU486 nor RTI001 induce AR dimerization and, similar to the AR antagonist Enzalutamide, were able to block R1881 mediated AR dimerization (Figs. 5b, c and S6A). To complement the NanoBiT assay, we employed a mammalian two-hybrid (M2H) interaction assay as an orthogonal approach and were unable to detect any AR/AR interactions at the concentrations of androgens that achieved maximal proliferative responses (lower than 0.1 nM). Similar to the NanoBiT format, only HD concentrations (>0.1 nM R1881) of androgens facilitated AR/AR interactions in the M2H assay (Supplemental Fig. S6B). Furthermore, neither RU486 nor the AR antagonist Enzalutamide induced AR dimerization (Supplemental Fig. S6B) as assessed in the M2H assay. Importantly, both RU486 and RTI001 were as effective as R1881 in inducing PCa cell proliferation, and this activity was inhibited by enzalutamide or a specific AR-directed Proteolysis Targeting Chimera (PROTAC) degrader (Fig. 5d, e). Reflecting their inability to facilitate AR dimerization, these ligands (RU486 and RTI001) were found to exhibit weak partial agonist activity on classical AR-responsive genes (i.e., PSA, FKBP5 or NDRG1). However, they functioned as full/super agonists with respect to the expression of LD mRNAs (i.e., E2F1 and FOXM1) and their corresponding proteins (Fig. 5f–h). Of particular importance, we determined that both compounds (a) are competitive AR antagonists (Fig. S6A, C–F), (b) disrupt N/C terminal interactions14,19, and (c) inhibit HD androgen-dependent activation of classical AR target genes but not LD androgen-dependent activation cell cycle genes (Fig. S6, C, E and F)14,19. Thus, these compounds recapitulate the biology/pharmacology of LD androgens by favoring the formation of AR monomers and confirm receptor oligomerization as a mechanism by which cells respond to different levels of androgens.

a Chemical structures for RU486 and RTI001. HEK293 cells were transfected in CFS-supplemented media with plasmids expressing AR fused to SmBiT or LgBiT. 48 h later, cells were treated with increasing concentrations of RU486 (b) or RTI001 (c) and compared to 10 nM R1881, at the time Nanoluciferase reagent was added, and read on plate reader. Data are shown as mean ± SD of a representative results from three independent experiments, n = 3 wells of cells. RLU: Relative Light Units. VCaP cells were plated in CFS media for 2 days and treated with increasing concentrations of RU486 (d) or RTI001 (e) alone or in the presence of Enzalutamide (Enz) or a PROTAC AR degrader and compared to LD R1881 (0.06 nM). Cell growth was assessed by DNA quantification at day 7. Data are plotted as mean ± SD of representative results from three independent experiments. n = 3 wells of cells. RFU relative fluorescent units. VCaP cells were plated in CFS-supplemented media for 2 days and treated with increasing concentrations of R1881, RU486 or RTI001 for an additional 24 h. The mRNA expression for PSA (f) and E2F1 (g) were assessed using qRT-PCR. Data are shown as mean ± SD as representative results from three independent experiments, n = 3 technical replicates. h VCaP cells were plated in CFS-supplemented media for 2 days and treated with either vehicle or indicated concentrations of R1881, RTI001 or RU486 for an additional 48 h. Whole cell extracts were probed for RB phosphorylation (pRB), E2F1, FOXM1, PSA, or β-Actin. Representative images are shown from n = 2 independent experiments. Source data are provided as a Source Data file.
AR-dependent activation of mTOR occurs in a non-genomic manner
Whereas monomeric AR induces the expression of mRNAs that encode proteins required for progression through the cell cycle, we made the unexpected finding that these responses were completely inhibited by cycloheximide at a concentration that did not block expression of mRNAs from the immediate-early (HD) primary target genes (e.g., PSA) (Fig. 6a, b). Thus, we concluded that the expression of the LD genes (i.e., E2F1) were secondary responses that required de novo protein synthesis. It has been suggested that mTOR activation is required for AR-dependent proliferative responses although the impact of dose on this process was never examined48. Using pS6 and p4EBP1 as biomarkers, we determined that mTOR was activated by both LD and HD androgens (Fig. 6c). These AR-dependent responses were inhibited by enzalutamide (Fig. S7A). Notably, RU486 and RTI001 were as effective as androgens in inducing mTOR activation, phosphorylation of RB, and the expression of E2F1 and FOXM1, implicating the AR monomer as the effector of these responses (Figs. 6c, and S7A). Unexpectedly, the ability of androgens to activate mTOR was insensitive to cycloheximide under conditions where protein synthesis was inhibited, highlighting non-genomic activities of AR (Figs. 6d, and S7B). Previous studies have concluded that mTOR activation occurred secondary to the AR-dependent induction of the expression of amino acid (AA) transporters, import of AAs, and activation of the RAGulator complex48. This conclusion is inconsistent with our data showing that mTOR activation is insensitive to cycloheximide treatment (Fig. S7B). Additionally, while insensitive to cycloheximide treatments, mTOR activation was detected prior to any measurable upregulation of classical AR-regulated proteins (e.g., NDRG1, FKBP5 or PSA) (Figs. S7C and 6e). Further, we used high-content imaging (Cellomics ArrayScan) to evaluate the subcellular localization of the AR in an unbiased manner in VCaP cells following treatment with increasing concentrations of androgens. As expected, the treatment of cells with HD androgens resulted in robust nuclear accumulation of AR (Fig. S7D). Under these experimental conditions, in agreement with our ChIP-seq analysis showing that AR was recruited to fewer genomic sites in response to LD androgens, we determined that LD androgens did not effectively stimulate nuclear translocation of AR (Fig. S7D). It has also been reported that AR can transiently (10–15 min) activate MAPK and AKT to reinforce the genomic actions of androgens49,50,51,52. We do not believe that this is related to the activation of mTOR that we observe as these rapid responses were only observed upon treatment with supraphysiological levels of agonists (Fig. S7E). Further, we demonstrate that the activation of mTOR in response to androgens occurs within ~4 h (Fig. S7C, D) and is maintained for 72 h (Fig. 6e). mTOR activation preceded the phosphorylation of RB and the increased expression of E2F1 and FOXM1 (Fig. 6e). Further, RB phosphorylation and the expression of E2F1 and FOXM1 mRNA and proteins were inhibited by the mTOR inhibitors Torin 2 or AZD8055. Inhibition of mTOR had little impact on the expression of PSA (Figs. 6f, and S7F–I). Whereas the mechanisms(s) by which androgens activate mTOR remain to be determined, it has recently been shown that AR can physically interact with mTOR in cells53,54. Regardless, these data indicate that androgens facilitate an AR monomer-dependent, non-genomic, activation of mTOR that results ultimately in RB phosphorylation and increased expression and translation of mRNAs encoding key cell cycle proteins (i.e., E2F1, FOXM1).

VCaP cells were plated in media supplemented with CFS for 2 days and treated for an additional 48 h with indicated concentrations of R1881 alone or in combination with cycloheximide (CHX). The mRNA expression for PSA (a) and E2F1 (b) were assessed using qRT-PCR. Data are shown as mean ± SD as representative results from three independent experiments, n = 3 technical replicates. c VCaP cells were plated in CFS-supplemented media for 2 days and treated with either vehicle or indicated concentrations of R1881, RTI001 or RU486 for an additional 48 h. Whole cell extracts were probed for RB phosphorylation (pRB), E2F1, FOXM1, PSA, phospho-S6 ribosomal protein (pS6), phospho 4EBP1 (p4EBP1) or β-Actin using western blot. d VCaP cells were treated with indicated concentrations of R1881 alone or in combination with cycloheximide (CHX) for an additional 24 h. Whole cell extracts were probed for RB phosphorylation (pRB), E2F1, FOXM1, PSA, phospho-p70 S6 Kinase (pS6K), phospho-S6 ribosomal protein (pS6), phospho 4EBP1 (p4EBP1) or β-Actin using western blot. e VCaP cells were treated with either vehicle or R1881 (0.06 nM or 10 nM) for indicated time. Whole cell extracts were probed for RB phosphorylation (pRB), E2F1, FOXM1, PSA, phospho-S6 ribosomal protein (pS6), or β-Actin using western blot. f VCaP cells were plated in media supplemented with CFS for 2 days and treated with indicated concentrations of R1881 alone (Veh) or in the presence of 100 nM Torin2 for 48 h. Whole cell extracts were probed for RB phosphorylation (pRB), E2F1, FOXM1, PSA, phospho-p70 S6 Kinase (pS6K), phospho-S6 ribosomal protein (pS6), phospho 4EBP1 (p4EBP1) or β-Actin using western blot. For c–f, representative images are shown from n = 3 independent experiments. Source data are provided as a Source Data file.
HD androgens suppress c-MYC expression to inhibit proliferation
The results of our studies revealed that both LD and HD androgens facilitate an AR monomer-dependent activation of mTOR; an activity required for RB phosphorylation and to increase the expression and translation of mRNAs encoding key cell cycle proteins (Figs. 6 and S7). However, despite mTOR activation, HD androgens inhibit the expression of mRNAs encoding proteins involved in the G1/S transition, a robust response that has been observed previously39,55,56,57. The mechanism(s) by which AR accomplishes this activity has not yet been resolved. Our RNA-seq analysis revealed that Hallmark c-MYC targets were upregulated in response to LD androgens but repressed in HD androgen-treated cells (Fig. 1g). Therefore, we wanted to determine the temporal impact of androgen levels on mTOR activation markers and on the expression of c-MYC, a primary oncogenic driver of PCa58,59. Interestingly, while both LD and HD androgens facilitate mTOR activation, only HD androgens repressed the expression of c-MYC suggesting that both mTOR activation and c-MYC are required to phosphorylate RB and to increase the expression and translation of mRNAs encoding key cell cycle proteins (Fig. 7a).

a VCaP cells grown in CFS-supplemented media were treated with vehicle, 0.1 or 10 nM R1881 for indicated time. b VCaP cells in CFS-supplemented media were treated with vehicle or R1881, RU486 or RTI001 as indicated. Whole cell extracts (WCE) were probed for pRB, E2F1, FOXM1, pS6K, pS6, p4EBP1, PSA, and β-Actin. c LNCaP cells in FBS-supplemented media were treated with increasing concentrations of either R1881 or AR antagonists. Bic: Bicalutamide, Enz: Enzalutamide or an AR degrader (PROTAC). d Castrated male NSG mice bearing LNCaP-AR tumors were administered either Vehicle (Veh, n = 12 mice), testosterone (T, n = 11 mice) or Enzalutamide (Enz, n = 11 mice). Data are plotted as mean ± SD and p values determined by two-way ANOVA followed by Tukey’s multiple comparison test (P < 0.0001). LNCaP (e) or VCaP (f) cells in FBS-supplemented media were treated for 48 h with Vehicle (Veh), 10 nM R1881 or AR antagonists. (Enz 10μM or PROTAC 2 μM). WCE were probed for AR, PSA, pRB, E2F1, FOXM1, c-MYC or β-Actin. LNCaP control (CTRL) or HD resistant (HDr) cells in FBS- (g) or in CFS (h) -supplemented media were treated with increasing concentrations of R1881. i LNCaP control (CTRL) or HD resistant (HDr) cells grown in FBS-supplemented media were treated for 48 h. with Vehicle (-) or 10 nM R1881 (+). WCE were probed for AR, PSA, pRB, E2F1, FOXM1, c-MYC and β-Actin. LNCaP control (CTRL) or LNCaP c-MYC (c-MYC) cells were plated in FBS (j) or CFS (k) -supplemented media and treated with increasing concentrations of R1881. LNCaP control (CTRL) or LNCaP c-MYC (c-MYC) cells in FBS (l) or CFS (m) -supplemented media and treated with increasing concentrations of R1881. WCE were probed for AR, PSA, pRB, E2F1, FOXM1, c-MYC and β-Actin. For A, B, E, F, I, L and M, representative images are shown from n = 3 independent experiments. For C, G, H, J, and K, Data are shown as mean ± SD of representative results from three independent experiments, n = 3 wells of cells. RFU: Relative Fluorescence Units. Source data are provided as a Source Data file.
Next, we wanted to determine the impact of AR dimerization on the expression of c-MYC. Interestingly, at doses that induce AR dimerization (Fig. 4c), androgens induce the expression of HD genes (i.e., PSA) and quantitatively inhibit c-MYC, E2F1, and FOXM1 expression and the phosphorylation of RB (Fig. 7b). Of note was the observation that the expression of c-MYC protein was eliminated within 4 h of the addition of HD androgens (Fig. 7a). Importantly, we also determined that AR modulators that fail to induce AR dimerization (i.e., RU486 and RTI001) were not able not repress c-MYC expression (Fig. 7b). Thus, AR dimerization is required to repress c-MYC levels.
As a follow-up study, we performed a comparative analysis of the inhibitory pharmacology of antiandrogens and HD androgens in cellular and animal models of PCa. This study revealed that AR agonists are significantly more efficacious in inhibiting PCa cell proliferation than even the most contemporary antiandrogen enzalutamide or an AR-directed VHL-PROTAC (Figs. 7c, and S8A). The proliferation of EnzR-VCaP, a cell line developed from VCaP tumors that progressed in castrated male mice treated with enzalutamide, was also more sensitive to inhibition with HD androgens than to AR antagonists (Fig. S8B). Importantly, it was also demonstrated that HD T inhibits the growth of LNCaP-AR cells when propagated as a xenograft (expressing 3 times the amount of AR when compared to the parental LNCaP and a model for castration resistant prostate cancer)60 (Fig. 7d). Follow-up studies revealed that while antiandrogens efficiently repressed the expression of AR targets such as PSA, they had minimal inhibitory effects on RB phosphorylation or on the expression of E2F1 and FOXM1 (Figs. 7e, f and S8C). Conversely, while HD androgens enhanced the expression of PSA, they were effective inhibitors of RB phosphorylation and E2F1 and FOXM1 protein expression. One of the most significant differences noted was that as opposed to HD androgen, antiandrogens failed to inhibit the expression of c-MYC in any cell model (Figs. 7e, f and S8C).
Given the primacy of c-MYC in AR pathobiology, we next probed the causal relationships between its expression and the ability of HD androgens to repress PCa proliferation. As a starting place for this study, we passaged PCa cells in the continued presence of HD androgens to the point of resistance. The resultant isogeneic variants HDr, while insensitive to treatment with HD androgens, remained dependent on androgens for proliferation and retained their sensitivity to Enz/PROTACs (Figs. 7g–i and S8D, E). Importantly, in HD-resistant cells, c-MYC expression is maintained and unresponsive to HD androgens (Fig. 7i). Further, RB phosphorylation and the expression of the cell cycle genes E2F1 and FOXM1 remain elevated in the HD androgen-treated cells (Fig. 7i). These results suggested that downregulation of c-MYC expression may be the primary mediator of the antiproliferative activity of HD androgens. To probe this relationship further, we engineered LNCaP cells that express c-MYC from a heterologous promoter and demonstrated that these cells were completely resistant to the anti-proliferative effects of HD androgens (Fig. 7j, k). Furthermore, treatment with HD androgens did not inhibit RB phosphorylation or the expression of E2F1 and FOXM1 (Fig. 7l, m) in these engineered cells. Together these data indicate that HD androgens facilitate AR dimerization, which mediates their antiproliferative activities through active suppression of c-MYC expression.
Discussion
Our work demonstrates that monomeric and dimeric/oligomeric AR are functionally distinct entities that regulate different signaling pathways to exhibit unique biological responses in cells. Considering this information, we propose a substantially revised model of AR action in which androgens activate and enable the release of AR from the HSP complex, but when fractional occupancy is low, the liganded receptor is predominately located in the cytoplasm as a monomer where it directly or indirectly activates the mTOR signaling pathway. Importantly, at doses of hormone that induce PCa cell proliferation and maximal upregulation of LD genes (e.g., E2F targets), only minimal AR nuclear translocation, chromatin AR binding, and alterations in chromatin architecture are observed (by ATAC-seq and ChIP-seq analysis). This suggests that the activity of AR on these target genes likely occurs in a non-canonical manner, with the monomeric AR facilitating a non-genomic activation of mTOR leading to increased RB phosphorylation and promoting cell proliferation. As hormone levels increase so too does the population of dimers which partition to the nucleus, interact with DNA, and subsequently increase the expression of classical AR target genes. However, HD androgens also initiate a negative feedback pathway which engages AR dimers in the active repression of c-MYC expression leading to RB1 dephosphorylation and downregulation of genes associated with the G1/S transition61, ultimately inhibiting cell proliferation. Thus, while both LD and HD androgens activate mTOR, only HD androgens repress c-MYC expression. It was recently reported that Rb loss is not required for HD androgen-mediated growth suppression, suggesting that MYC downregulation is sufficient to halt cell cycle progression, independent of Rb status61. The mechanisms by which the AR dimer actively suppress c-MYC expression remain to be determined, but considering the findings of others it is possible that the dimer engages the inhibitory DREAM complex to accomplish this activity61. Together, these findings explain how the same hormone acting through the same receptor can manifest different biological responses in the same cell and how prostate tumors exhibit non-linear responses to androgens.
Previous studies have shown that AR and mTOR can physically interact within the nucleus, leading to the reprogramming of mTOR-dependent metabolic gene networks53. However, this interaction has only been observed in cells treated with HD androgens, whereas in the presence of LD androgens, the compartmentalization of AR/mTOR remains cytoplasmic. While AR is required for PCa cell proliferation in response to LD androgens, ChIP-seq and ATAC-seq analysis, together with studies of AR compartmentalization suggest that nuclear mTOR/AR complexes are unlikely to mediate proliferation in response to LD androgens. Rather, our data suggest that androgens facilitate an AR monomer-dependent, non-genomic activation of mTOR, ultimately promoting cell proliferation. Further investigation will be required to define the specific mTOR complex (C1 or C2) required for the proliferative response to androgens. Several studies have proposed that the oligomeric state of the Glucocorticoid Receptor (GR) enables it to exhibit distinct functions in the nucleus likely reflecting its ability to interact with GRE-half sites and palindromic response elements62,63,64. This is in contrast with our studies with AR where we have shown that monomeric and dimeric receptors exhibit distinct functions in different cell compartments. It has been shown recently that unliganded cytoplasmic GR can interact with KRAS and inhibit downstream PI3K-AKT and MAPK pathways in the cytoplasm. The extent to which this can be attributed to monomeric vs. dimeric GR complexes remains to be determined65. How and if other steroid hormones take advantage of receptor oligomerization to elicit diverse biological responses to alterations in hormone levels remains to be explored. We have determined that cancer cells do respond differently to different levels of progestins although the role of oligomerization in this activity has not been determined66,67.
The revised model of AR action we propose has significant implications with respect to strategies currently used to inhibit AR action in PCa. Notably, it suggests that the specific inhibition of the activity of monomeric AR is the optimal approach to block the pathobiological actions of androgen in PCa. The current approaches used to target AR signaling (competitive antagonists, PROTACs, CYP17 inhibitors, and GnRH agonists/antagonists) have the potential to effectively inhibit monomeric AR if they achieve absolute inhibition of the receptor. However, any monomeric AR remaining will be free to engage in the regulation of LD biology. All existing AR-directed PCa therapies will disrupt the favorable activities of the dimeric receptor and would likely also increase the propensity for transdifferentiation of PCa cells to neuroendocrine prostate cancer68,69. Our findings suggest that the paradoxical approach of facilitating AR dimerization and exploiting the hardwired negative feedback pathway on processes required for cell proliferation may be a more effective way to inhibit androgen action in PCa (Fig. 8). Recently, an antagonist was described that binds to the AR dimer interface suppressing AR signaling70. Although this inhibitor targets the AR dimerization interface, it likely impacts proliferation of PCa cells through mechanisms distinct from disruption of AR dimer activities. Indeed, by binding to the dimer interface, these ligands could disrupt essential structural features of the monomeric AR (e.g., specific coregulator recruitment or altering AR conformation), thereby interfering with its ability to drive proliferation potentially inhibiting mTOR activation. Interestingly, while the AR N/C interactions were described to be dispensable for normal development in male mice71, our studies indicate that this interaction is important for AR/AR dimerization, the regulation of classical ARE-containing genes, and the downregulation of c-MYC expression. This is supported by our observation that drugs that do not induce N/C interactions fail to induce AR/AR dimerization and exhibit weak agonist activity on classic AR target genes (e.g., PSA). Further, such drugs fail to downregulate the expression of c-MYC.

Upon binding an androgen AR undergoes a conformational change that enables its release from an inhibitory heat-shock protein complex (HSP). In the presence of castrate levels of androgens, AR fractional occupancy is low, and the receptor is predominately located in the cytoplasm as a monomer. Monomeric AR activates the mTOR signaling pathway enabling cell proliferation. As the levels of hormones increase to eugonadal levels, the fractional occupancy of AR increases and the population of dimers which partition to the nucleus rise, interact with DNA, and enhance the expression of classical AR target genes. Simultaneously, AR dimers actively repress c-MYC expression to initiate a negative feedback pathway leading to RB1 dephosphorylation and the repression of genes associated with the G1/S transition. This model also predicts that when androgens levels are low, androgen deprivation therapy (ADT) consisting of anti-androgens, or the ligands directed degraders (LDD)s will promote castration resistant neuroendocrine prostate disease. Conversely, when androgens levels are high, ADT will favor the formation of the monomeric AR and drive tumor growth.
Although HD androgens effectively inhibit RB phosphorylation and the expression of cell cycle genes in both VCaP and LNCaP cells, repression is more robust in VCaP cells, which express higher levels of AR (Figs. S1A, B, and S6E and F). As the ability of HD androgens to inhibit PCa proliferation is influenced by AR expression level, this activity may be more robust in PCa cells/tumors where receptor levels are elevated and engaged in the regulation of activities that do not occur in normal cells72. Several studies have explored the therapeutic utility of HD androgens (monthly bolus of testosterone cypionate) in patients with metastatic PCa, a strategy called Bipolar Androgen Therapy (BAT)32,33,34. Indeed, the results of the recent TRANSFORMER study indicated that BAT was as effective as Enzalutamide in patients who had progressed on the CYP17 inhibitor abiraterone33. It is notable that high AR activity, rather than high AR expression per se, has been identified as a predictive biomarker of response to this treatment and transcriptional downregulation of AR has emerged as a mechanism of resistance to HD androgens in a subset of patients33. Our data suggests that the BAT protocol does not optimally leverage the mechanisms by which HD androgens suppress proliferation as blood levels of androgens decline in the second half of the BAT treatment cycle likely favoring the formation of monomeric AR to potentially restore proliferative responses. Thus, there appears to be an unmet medical need for an AR modulator which favors the formation of AR dimers, and which is devoid of the liabilities of existing androgen agonists in the liver, cardiovascular, and central nervous systems73,74,75,76. Beyond the realm of cancer, this revised model of AR action/pharmacology has implications with respect to the strategies that are currently used to treat hypogonadism in males, and those used to treat the climacteric conditions associated with menopause in females. The discordance between dose and proliferation noted for androgens also begs a reevaluation of the established approaches used to evaluate the activity of chemicals in the environment as potential disrupters of androgen signaling.
Methods
Animal studies
All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, 8th ed., and approved by the Duke University Institutional Animal Care and Use Committee (AUP# A220-21-11 and A278-18-12). Animals were euthanized when tumor volume exceeded the 2 cm3 total volume limit imposed by the Duke IACUC. Six-week-old male NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were obtained from Jackson Laboratories (Bar Harbor, ME) and were maintained under specific pathogen-free, temperature- and humidity-controlled conditions, with a 12-h light/12-h dark schedule. For LNCaP and VCAP studies, mice were castrated, assigned to groups (n = 10) and implanted subcutaneously (sc) with placebo pellets or with pellets (90-day duration) containing increasing doses of either (T, NA-151; 1.25, 2.5, 5, 10 and 20 total dose mg/pellets) or 5a-dihydrotestosterone (DHT, NA-161; 0.1, 0.25, 0.5, 1, 2 and 5 total dose mg/pellets) (Innovative Research of America, Sarasota, FL). 10 days later, mice were injected with 3 × 106 LNCaP or 1 × 106 VCaP Cells in 50% matrigel (BD Matrigel Matrix, BD Biosciences, San Jose, CA) sc into the right flank. Tumor size was measured with calipers 3 times weekly and tumor volume was calculated using the formula volume = width2 × length/2. For the LNCaP-AR xenograft study, male NSG mice (in house colony, Duke Cancer Institute, Durham, NC) were castrated at 6 weeks of age, 10 days before injection of 3 × 106 LNCaP-AR cells sc into the flank. When tumor volume reached ∼0.15–0.2 cm3, mice were randomized (n = 10–12) to receive vehicle (10% DMSO/1% CMC/0.1% Tween 80; po qd), Enzalutamide (30 mg/kg; po qd) or testosterone (50 mg/kg; sc qw in sesame oil, vehicle po qd). Group size per treatment arm included: Vehicle n = 12, Enzalutamide n = 10 and Testosterone n = 11.
Reagents
LNCaP cells were maintained in RPMI supplemented with 8% FBS. VCaP, EnzR-VCaP, HEK293, or CV-1 cells were maintained in DMEM (8% FBS). All cell lines were obtained from ATCC which uses short tandem repeat (STR) DNA profiles for authentication. None of the cell lines used for these studies are listed in the database of commonly misidentified cell lines maintained by ICLAC. All cell lines tested negative for mycoplasma. Stable LNCaP cell lines expressing c-MYC were generated using pCDH-puro-cMyc (addgene, plasmid #46970). Torin2 and AZD8055 were purchased from Selleckchem. RU486 (Mifepristone), Enzalutamide, and bicalutamide were purchased from Cayman Chemical Company (≥98% purity). RTI001 was a gift from E. Cook (Research Triangle Park, NC)44, Antibodies to AR (441, SC-7305, 1:10,000) and FKBP51 (D-4, sc-271547, 1:50,000) were purchased from Santa Cruz Biotechnology. Antibodies for β-Actin (AC-15, A5441, 1:800,000) and Anti-Puromycin (MABE343, 12D10, 1:300,000) was purchased from Sigma. Antibodies for Phospho-Rb (Ser807/811; D20B12, #8516, 1:50,000), Phospho-p70 S6 Kinase (Thr389; 1A5, #9206, 1:2000), Phospho-S6 Ribosomal Protein (Ser235/236; D57.2.2E, #4858, 1:4000) and Phospho-4E-BP1 (Ser65; #9451, 1:1000), c-MYC (D84C12, #5605, 1:2000), PSA (D11E1, #2475, 1:20,000), NDRG1 (#5196, 1:1000), FOXM1 (D3F2B, #20459, 1:2000) and Vinculin (E1E9V, #13901, 1:160,000) were purchased from Cell Signaling. E2F1 antibody (KH95/E2F, #554213, 1:1000) was purchased from BD Biosciences. FuGENE HD Transfection Reagent and furimazine were purchased from Promega.
Proliferation assay
LNCaP (4000 cells/well) and VCaP or EnzR-VCAP Cells (10,000 cells/well) were seeded in 96-well plates containing media supplemented with 8% fetal bovine serum (FBS) or charcoal-stripped fetal bovine serum (CFS). The following day for FBS or 2 days later when in media supplemented with CFS, the cells were treated with test compounds as indicated. Following a seven-day incubation, media was removed by inversion, and plates were frozen overnight at −80 °C. Plates were thawed at room temperature and 100 μl ddH2O was added to each well. Plates were incubated at 37 °C in non-CO2 incubator for 1 h. and then frozen at −80 °C overnight. Plates were thawed to room temperature and 100 μl TNE buffer (NaCl, Tris, EDTA) + Hoechst dye (1.0 mg/ml, 1:500) was added to each well. Cell proliferation was quantified by measuring DNA content using Hoechst dye77. Fluorescent signal was measured at 460 nm. Each sample was evaluated in triplicate, and the results from a representative experiment are shown. Results are expressed as relative fluorescence ± SD (n = 3 wells of cells).
RNA isolation and real-time PCR
LNCaP cells (130,000 cells/well) were seeded in 6-well plates in RPMI 1640 (8% CFS). For VCaP cell line (300,000 cells/well), cells were seeded in 6-well plates in DMEM (8% CFS). 48 h later, cells were treated with ligand for an additional 24 h and total RNA were isolated using the Aurum™ Total RNA Mini-Kit according to the manufacturer’s instructions (Bio-Rad). RNA quality and quantity were determined using a Nanodrop 1000 (Thermo Fisher) and total RNA (1 µg) was reverse transcribed to cDNA using iScript™ cDNA synthesis Kit (Bio-Rad). The resulting cDNA was diluted 1:20 with water for use in the qPCR analysis. qPCR was performed using the Bio-Rad SYBR green supermix with 0.2 M of each forward and reverse primer and 2.25 µl of diluted cDNA in a total reaction volume of 6 µl. PCR amplification was carried out using the Bio-Rad iQ4 or the CFX384 qPCR system. All primers used in the study were tested to have PCR efficiency between 100 ± 10% and span intron/exon boundaries when possible. Gene expression levels were first normalized to an internal control 36B4 or GAPDH, and then to control conditions. Each sample was performed in n = 3 technical replicate and the results from a representative experiment are shown. Gene-specific primers were purchased from Eton Bioscience Inc.
36B4 (For: GGACATGTTGCTGGCCAATAA, Rev: GGGCCCGAGACCAGTGTT)
GAPDH (For: ACAGTCCATGCCATCACTGCCACCCAGAAG; Rev: CAGTGAGCTTCCCGTTCAGCTCAGGGATGA)
PSA (For: CCTCCTGAAGAATCGATTCC; Rev: AGGTCCACACACTGAAGTT)
E2F1 (For: ACGTGACGTGTCAGGACCT; Rev: GATCGGGCCTTGTTTGCTCT)
FKBP5 (For: CGGAGAACCAAACGGAAAGG; Rev: CTTCGCCCACAGTGAATGC)
CDC6 (For: TGAATGGCCATGGCTAAGCA; Rev: GGAAGTTCAACAGCTGTGGC)
RNA-seq analysis
RNA-seq data were checked for quality and trimmed using FastQC (v0.11.9)78, MultiQC (v1.10.1)79 and Trimmomatic (v0.39)80; trimmed reads shorter than 36 bp were removed. Reads were aligned to the reference genome (GRCh38) and annotated to gene features using the STAR aligner (v2.7.8a)81. The reference genome and annotation files were obtained from GENCODE (Release 38)82. Read- and base-level mapping quality was checked using STAR output and Picard Toolkits (v2.23.8)83. Tests of differential gene expression were performed using R (v4.2.1)84 and the package DESeq2 (v1.36.0)85. Path-analyses were performed using fgsea (v1.22.0)86 with Hallmark87 gene sets.
nCounter NanoString gene expression profiling
VCaP cells were plated in a 96-well plate at a density of 50,000 cells/well in 5%CFS-supplemented media. 24 h later, cells were treated with either vehicle (Veh), LD (0.03 nM) or HD R1881 (0.3 nM) for an additional 24 h. Cells were lysed using Qiagen buffer RLT (15 μL/well) and total RNA extracted. The assay was performed in duplicate in accordance with the nCounter XT CodeSet Gene Expression assay (NanoString assay) manufacturer’s manual (NanoString Technologies).
Sample preparation for ATAC-seq
VCAP or LNCaP cells were plated in CFS containing media for 48 h and treated with vehicle or increasing concentrations of R1881 for 24 h. Cells were then washed with PBS, trypsinized, and washed again with PBS in preparation for transposition as described in Buenrostro et al.88. The OMNI protocol was followed for ATAC-seq89, but briefly, harvested cells were spun down at 500 × g for 5 min at 4 °C to pellet the cells. Supernatant was aspirated and pellets resuspended in 50 μL cold resuspension buffer (RSB) (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2) containing 0.1% NP40, 0.1% Tween-20 and 0.01% Digitonin. Cell pellets were resuspended by gentle pipetting with wide bore tips and incubated on ice for 3 min. After 3 min, 1 mL of cold RSB containing 0.1% Tween 20 was added to tubes, and tubes inverted 3 times to mix. Tubes were spun at 500 × g for 10 min at 4 °C. Supernatant was aspirated, and pellets resuspended in 25 μL 2× TD Buffer (Illumina Cat #FC-121-1030, 20 mM Tris-HCl pH 7.6, 10 mM MgCl2, 20% Dimethyl Formamide). 2 μL were taken out from each sample to assess nuclei quality and lysis efficiency, as well as count the total number of nuclei. 60,000 nuclei from each condition were then put into a new tube and volume brought up to 25 μL with 2X TD Buffer. 25 μL of transposition reaction mix (per reaction: 2.5 μL) Tn5 Transposase (Illumina Cat #FC-121-1030 + 16.5 μL 1X PBS + 0.5 μL10% Tween-20 + 0.5 μL 1% Digitonin + 5 μL Nuclease Free H2O) was then added to each tube, totaling 50 μL per sample. Reaction mixtures were gently pipetted and then incubated at 37 °C for 30 min in a thermomixer shaking at 1000RPM. After 30 min, enzymatic reaction was stopped, and samples purified using the Qiagen MinElute Kit (#28204). Transposed DNA was eluted in 10 μL Elution Buffer (10 mM Tris buffer, pH 8). To make ATAC-seq libraries, purified DNA was then amplified via PCR with the following reaction conditions: 10 μL Transposed DNA + 12.5 μL Nuclease Free H2O + 2.5 μL 25 μM Customized Nextera PCR Primer-1 + 2.5 μL 25 μM Customized Nextera PCR Primer-2 + 25 μL NEBNext Ultra II Q5 Master Mix (Cat # M0544S), total 50 μL per reaction. Each sample has a unique combination of Primer 1 and Primer 2 to double-index the samples. The following PCR reaction was then run: (1) 72 °C, 5 min, (2) 98 °C, 30 s, (3) 98 °C, 10 s, (4) 63 °C, 30 s, (5) 72 °C, 1 min, (6) steps 3–5, were repeated 4× times, (7) hold at 4 °C. Next a qPCR side reaction was performed to monitor and stop amplification prior to saturation to reduce GC and size bias. The side reaction consisted of the following components: 5 μL 5 cycles PCR amplified DNA + 4 μL Nuclease Free H2O + 0.5 μL 50 μM Customized Nextera PCR Primer 1 + 0.5 μL 50 μM Customized Nextera PCR Primer 2 + 0.06 μL 100× SYBR Green I + 10 μL NEBNext Ultra II Q5 Master Mix, totaling 20 μL per sample. The following qPCR cycling conditions were used: (1) 98 °C, 30 s, (2) 98 °C, 10 s, (3) 63 °C, 30 s, (4) 72 °C, 1 min, (5) steps 2–4, repeated 19× times, (6) Hold at 4 °C. Once the side reaction was completed, the amplification curves of the samples were analyzed to determine the additional number of cycles needed for the remaining 45 μL PCR reactions. This was determined by (1) Plot linear Rn vs. Cycle, (2) Set 5000 RF threshold, (3) Calculate the # of cycles that corresponded to 1 ⁄4 of maximum fluorescent intensity. Then the remaining 45 μL PCR reactions were run to their corresponding correct number of cycles. They were cycled as follows: (1) 98 °C, 30 s, (2) 98 °C, 10 s, (3) 63 °C, 30 s, (4) 72 °C, 1 min, (5) steps 2–4 repeated, x predetermined times, (6) Hold at 4 °C. After PCR reactions were complete, the amplified library was purified using a 1:1 ratio of Ampure XP beads according to manufacturer instructions. The purified library was eluted in 20 μL Elution Buffer (10 mM Tris Buffer, pH 8). An aliquot of each sample was run on a TapeStation to check for transposition efficiency. All samples were barcoded (24 barcodes) and combined into pools and sequenced on a NovaSeq 6000 sequencer at the Duke Sequencing and Genomic Technologies shared resource.
Sample preparation for ChIP-seq
VCAP or LNCaP cells were plated in CFS containing media for 48 h and treated with vehicle or increasing concentrations of R1881 for 24 h. Cells were plated at the density required to obtain at least 5 million cells at time of collection. Media was then aspirated and 15 mL of fixation buffer (50 mM Hepes-KOH ph7.5, 100 mM NaCl, 1.0 mM EDTA, 0.5 mM EGTA, 3.7% formaldehyde) was added for exactly 10 min. Fixation was quenched with 125 mM glycine for 5 min. Fixation buffer was removed, and cells were scraped into 2 mL ice-cold wash buffer PBS plus 1X detergent (Active Motif (catalog number 37517)). 3 mLs of wash buffer was added for a total of 5 mL and cells were centrifuged for 5 min at 1250 g (4 °C). Supernatant was aspirated and cell pellet was resuspended in 1 mL of wash buffer, 4 mL of wash buffer was added, and cells were centrifuged for 5 min at 1250 × g (4 °C). Supernatant was carefully aspirated, and cell pellet was flash frozen in liquid nitrogen (LN2) and stored at −80 °C. For sonication, cells were thawed and resuspended in ice cold LB1 Buffer (50 mM Hepes-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) containing 1× Halt™ 100X Protease and Phosphatase Inhibitor Cocktail (ThermoFisher #78446) and rotated at 4 °C for 10 min. Cells were then centrifuged, and the pellets resuspended in LB2 Buffer (10 mM Tris-HCL pH 8, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) containing 1× Halt™ 100X Protease and Phosphatase Inhibitor Cocktail. Cells were then centrifuged, pellets were resuspended in ice cold LB3 Buffer (10 mM Tris-HCl pH 8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Sodium Deoxycholate, 0.5% N-laurylsarcosine) containing 1× Halt™ 100X Protease and Phosphatase Inhibitor Cocktail. This nuclear resuspension was transferred into Covaris microtube 130 AFA Fiber pre-Slit snap-cap tubes (Covaris #520045) at a maximum concentration of 3 million cells per 130 µL and sonicated using the following parameters on a Covaris ME220 according to the manufacturer’s recommendations (Duration (s) = 360, Peak Power = 75, Duty Factor = 5.0, Cycles/Burst = 1000). Following sonication, a 10 µL aliquot was removed for analysis of resulting DNA size and concentration. Briefly, 10 µL of sonicated chromatin was incubated with 20 µg RNase A (Thermo Scientific #EN0531) in TE pH 8 for 30 min at 37 °C in a thermal cycler. 20 µg of Proteinase K (Thermo Scientific #EO0491) in Active Motif Elution Buffer AM4 (Active Motif #103926) was then added and the sample was incubated for 30 min at 55 °C followed by 90 min at 65 °C in a thermal cycler. Samples were then cooled to room temperature and DNA was purified using SPRIselect beads (Beckman #B23318) at a bead:sample ratio of 1.4:1 and eluted in 10 µL of 10 mM Tris, 0.1 mM EDTA. Chromatin Immunoprecipitation was then carried out using thawed chromatin corresponding to 1 million cells diluted with 200 µL in Active Motif ChIP Buffer (Active Motif #37516) and incubated with pre-clearing Protein G agarose beads prepared according to Active Motif’s Low Cell ChIP kit (Active Motif #53084). After pre-clearing the chromatin, agarose beads were pelleted, and cleared chromatin transferred to a new microcentrifuge tube containing 4 µg of antibody specific to the target protein (AR: Abcam ab236225). IPs were then incubated overnight on a rotator at 4 °C. Next, Protein G Agarose beads prepared the previous day according to Active Motif’s Low Cell ChIP kit were added to the antibody-bound chromatin and incubated for 4 h on a rotator at 4 °C. IPs were then transferred to ChIP filtration columns, washed, and eluted in 80 µL according to Active Motif’s Low Cell ChIP kit and DNA purified using SPRIselect beads at a bead:sample ratio of 1.4:1. After SPRI cleanup, ChIP samples and reverse crosslinked input DNA was made into sequencing Illumina libraries using Swift NGS 2S Plus (Swift Biosciences #21096) and indexed using Single Indexed Adapters with MIDs (Swift Biosciences #279384). ChIP and input libraries were sequenced on either an Illumina HiSeq 4000 or a NovaSeq 6000 using paired-end, dual-indexed reads (38 bp read1 x 38 bp read2 x 8 bp index1 x 8 bp index2) to a minimum depth of 30 million reads.
ATAC-seq/ChIP-seq analysis
ATAC-seq and ChIP-seq data were checked for quality and trimmed using tools previously described78,79,80. Reads were aligned to the reference genome (GRCh38) obtained from GENCODE (Release 38)82 using the BWA aligner (v0.7.17-r1188)90. Reads that were unaligned, multi-mapped, failed platform quality checks or had mapping quality less than 30 were removed using Samtools (v1.12)91. Duplicate reads were identified by MarkDuplicates program (v2.23.8) from Picard toolkit (v4.2.2.0)83 and were removed using Samtools. Reads falling into curated blacklisted regions92 were detected and removed using Bedtools (v2.29.2)93. Peak detection was performed using MACS2 (v2.2.7.1)94. Data were evaluated using ENCODE standards. Differential binding analyses were performed using the Bioconductor95 package DiffBind (v3.6.5)96,97 with R statistical environment (v4.2.1)84, and motif analyses were performed using the findMotifsGenome program in the Homer suite98. Peaks were annotated to gene features obtained from GENCODE (Release 38)82 using the annotatePeaks program in the Homer suite98, and nearest genes within 25 kb upstream and downstream of the peaks were used for gene set enrichment analyses using fgsea (v1.22.0)86. Peak signal profiles in bigwig format were generated and visualized as heatmaps using Deeptools suite (v3.5.1)99.
Immunoblot analysis
Cells were plated in appropriate media supplemented with 8% FBS or 8% charcoal dextran treated FBS (CFS). Following 24 h in FBS or 48 h when in CFS, cells were then treated as indicated for an additional 48 h. Whole-cell extracts were prepared using RIPA buffer and protease inhibitors: (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.05% SDS, 5 mM EDTA, 50 mM NaF, 15 mM Na-pyrophosphate, 10 mM β-glycerophosphate, 2 mM Na-orthovanadate, 1× protease inhibitor cocktail). Cleared whole cell extracts were analyzed by the Bradford assay and 20 μg of protein per sample were resolved by SDS-PAGE (10% polyacrylamide gels), transferred to PVDF membranes, and detected by western blot using the following antibodies: AR, pS6K, pS6, p4EBP1, pRB, E2F1, FOXM1, c-MYC, PSA, FKBP5, NDRG1, and β-Actin or vinculin as loading controls. Uncropped and unprocessed scans of blots are provided in the Source Data file.
NanoLuc Binary Technology (NanoBiT)
One day prior to transfection, HEK293T cells were seeded in a 10 cm dish containing media supplemented with 8% CFS at a density of 5 × 106 cells. The following day, AR-SmBiT and LgBiT-AR expression vectors were added to Fugene diluted in Opti-MEM (Gibco), incubated for 30 min at room temperature, and added to the cells drop-wise. 24 h post transfection, the cells were trypsinized and seeded at the density of 50,000 cells per well in 96-well white cell culture plates containing media supplemented with 8% CFS. The next day, luminescence was measured using a Nano-Glo® Live Cell Assay System (Promega), and drugs were added immediately after the addition of luminescence reagent.
High content imaging
VCaP Cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100, and stained for AR (1:400, N-20, Santa Cruz) and counterstained for DNA (DAPI, Sigma) and F-actin (rhodamine Phalloidin, Thermo Fisher Scientific). Stained cells were imaged and analyzed with a Cellomics ArrayScan VTI HCS system. 20 fields per well of a 24-well plate were imaged at 20× magnification and analyzed using the Compartmental Analysis Bioapplication. First, images were collected by autofocusing on nuclear staining in channel 1. Cells were then identified in channel 1, indicated as valid object count (VOC). Nuclear:Cytoplasmic ratio of AR staining was determined by measuring channel 2 signal within the nuclear mask identified in channel 1 versus the cytoplasmic area, which was approximated by extending 2 pixels outside of the nuclear mask. Experiments were performed in triplicate and repeated five times.
Reporter gene assay
CV1 cells were seeded into 96-well cell culture plates and transfected with Lipofectin (Invitrogen) according to the manufacturer’s protocol. DNA mixture consisted of VP16-AR full-length, GAL4-DBD-AR 507–919, 5xGal4Luc3 reporter plasmid, and Renilla-Luc (for assessing transfection efficiency and toxicity). Following an overnight incubation, cells were treated with ligands for 24 h. Cells were lysed and luciferase activity was quantified using Dual Luciferase Reagent (DLR).
Statistical analyses
RNA-seq normalization and differential expression was carried out using the DESeq Bioconductor package with the R statistical programming environment. False discovery rate (FDR) was calculated to control for multiple hypothesis testing. Statistical significance was set at an FDR < 0.05.
NanoString data were analyzed using the R package ‘NanoStringNorm’ and normalized for technical assay variation using the CodeCount method for NanoStringNorm with geometric means. Samples were normalized for RNA content using the NanoStringNorm SampleContent method with geometric mean and the housekeeping genes (19 genes). The statistical effect of R1881 concentrations were analyzed and visualized using the statistical programming language R. Log2 fold changes of genes in heatmaps were calculated based on difference between the R1881 LD or HD samples and the average of the vehicle samples.
For xenograft studies, using the sample size and the power function in JMP statistical software (SAS Institute, Inc, Cary, NC.), it was estimated that a group size of 10–12 per treatment arm would be required to reliably detect with 80% confidence a statistically relevant (P < 0.05) change of 30% given the anticipated 15% variability for the tumor models used in these studies (α = 0.05, s.d. = 0.15, confidence of 0.8, s/delta of 0.3). These estimates were based on one way ANOVA followed by the Student Newman–Keul’s test. This group size is in accordance with current literature in the field. The investigator and personnel were not blinded during these studies. Significant differences (p < 0.05) in tumor growth over time was analyzed by 2-way ANOVA of repeated measures (without Geisser-Greenhouse correction), followed by Tukey’s multiple comparison between mean values of all groups at each time point. Data presented are average tumor volume ± standard error of the mean (s.e.m.) at each data point. For those animals that died prior to completing treatment courses (no more than 1 per group), recorded data was excluded from graphing and statistical analyses.
For in vitro studies, standard deviation (SD) is reported in figure legends for technical replicates from representative experiments performed in duplicates or triplicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data needed to evaluate the conclusions from this work are available in the main text or the supplementary materials. The RNA-seq, ATAC-seq and Chip-seq data generated in this study have been deposited in the GEO database under the accession number SuperSeries GSE247593. Source data are provided with this paper.
Code availability
The code to reproduce the results from the RNA-seq, ATAC-seq and Chip-seq data is available through the project’s source code repository (https://gitlab.oit.duke.edu/dcibioinformatics/pubs/mcdonnell-lab-androgens-dose-2023).
References
Davey, R. A. & Grossmann, M. Androgen receptor structure, function and biology: from bench to bedside. Clin. Biochem. Rev. 37, 3–15 (2016).
Notelovitz, M. Androgen effects on bone and muscle. Fertil. Steril. 77, S34–S41 (2002).
Khosla, S. & Monroe, D. G. Regulation of bone metabolism by sex steroids. Cold Spring Harb. Perspect. Med. 8, a031211 (2018).
Navarro, G., Allard, C., Xu, W. & Mauvais-Jarvis, F. The role of androgens in metabolism, obesity, and diabetes in males and females. Obesity 23, 713–719 (2015).
Morford, J. J., Wu, S. & Mauvais-Jarvis, F. The impact of androgen actions in neurons on metabolic health and disease. Mol. Cell Endocrinol. 465, 92–102 (2018).
Vanderschueren, D. et al. Sex steroid actions in male bone. Endocr. Rev. 35, 906–960 (2014).
Westaby, D. et al. A new old target: androgen receptor signaling and advanced prostate cancer. Annu. Rev. Pharm. 62, 131–153 (2022).
Penning, T. M. New frontiers in androgen biosynthesis and metabolism. Curr. Opin. Endocrinol. Diabetes Obes. 17, 233–239 (2010).
Okeigwe, I. & Kuohung, W. 5-Alpha reductase deficiency: a 40-year retrospective review. Curr. Opin. Endocrinol. Diabetes Obes. 21, 483–487 (2014).
Swerdloff, R. S., Dudley, R. E., Page, S. T., Wang, C. & Salameh, W. A. Dihydrotestosterone: biochemistry, physiology, and clinical implications of elevated blood levels. Endocr. Rev. 38, 220–254 (2017).
Chatterjee, P. et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Invest. 129, 4245–4260 (2019).
Kumar, R., Sena, L. A., Denmeade, S. R. & Kachhap, S. The testosterone paradox of advanced prostate cancer: mechanistic insights and clinical implications. Nat. Rev. Urol. https://doi.org/10.1038/s41585-022-00686-y (2022).
Grino, P. B., Griffin, J. E. & Wilson, J. D. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 126, 1165–1172 (1990).
Norris, J. D. et al. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem. Biol. 16, 452–460 (2009).
Chang, C. Y. & McDonnell, D. P. Androgen receptor-cofactor interactions as targets for new drug discovery. Trends Pharm. Sci. 26, 225–228 (2005).
Dehm, S. M. & Tindall, D. J. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol. Endocrinol. 21, 2855–2863 (2007).
Heery, D. M., Kalkhoven, E., Hoare, S. & Parker, M. G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387, 733–736 (1997).
Tzukerman, M. T. et al. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol. Endocrinol. 8, 21–30 (1994).
Kazmin, D. et al. Linking ligand-induced alterations in androgen receptor structure to differential gene expression: a first step in the rational design of selective androgen receptor modulators. Mol. Endocrinol. 20, 1201–1217 (2006).
Chang, C. et al. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol. Cell Biol. 19, 8226–8239 (1999).
Paige, L. A. et al. Estrogen receptor (ER) modulators each induce distinct conformational changes in ER alpha and ER beta. Proc. Natl Acad. Sci. USA 96, 3999–4004 (1999).
Wijayaratne, A. L. et al. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology 140, 5828–5840 (1999).
He, B. et al. An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein (CHIP). J. Biol. Chem. 279, 30643–30653 (2004).
He, B., Bowen, N. T., Minges, J. T. & Wilson, E. M. Androgen-induced NH2- and COOH-terminal Interaction Inhibits p160 coactivator recruitment by activation function 2. J. Biol. Chem. 276, 42293–42301 (2001).
He, B. et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 16, 425–438 (2004).
He, B., Kemppainen, J. A., Voegel, J. J., Gronemeyer, H. & Wilson, E. M. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH(2)-terminal domain. J. Biol. Chem. 274, 37219–37225 (1999).
He, B., Kemppainen, J. A. & Wilson, E. M. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 275, 22986–22994 (2000).
He, B., Lee, L. W., Minges, J. T. & Wilson, E. M. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J. Biol. Chem. 277, 25631–25639 (2002).
He, B., Minges, J. T., Lee, L. W. & Wilson, E. M. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J. Biol. Chem. 277, 10226–10235 (2002).
He, B. & Wilson, E. M. Electrostatic modulation in steroid receptor recruitment of LXXLL and FXXLF motifs. Mol. Cell Biol. 23, 2135–2150 (2003).
Yu, X. et al. Structural insights of transcriptionally active, full-length androgen receptor coactivator complexes. Mol. Cell 79, 812–823.e814 (2020).
Mohammad, O. S. et al. Supraphysiologic testosterone therapy in the treatment of prostate cancer: models, mechanisms and questions. Cancers 9, 166 (2017).
Denmeade, S. R. et al. TRANSFORMER: a randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castration-resistant metastatic prostate cancer. J. Clin. Oncol. 39, 1371–1382 (2021).
Cai, C. & Balk, S. P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 18, R175–R182 (2011).
Mohler, J. L. et al. The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 10, 440–448 (2004).
Montgomery, R. B. et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 68, 4447–4454 (2008).
Sharifi, N. The 5alpha-androstanedione pathway to dihydrotestosterone in castration-resistant prostate cancer. J. Investig. Med. 60, 504–507 (2012).
Nyquist, M. D. et al. Selective androgen receptor modulators activate the canonical prostate cancer androgen receptor program and repress cancer growth. J. Clin. Invest. 131, e146777 (2021).
Nyquist, M. D. et al. Molecular determinants of response to high-dose androgen therapy in prostate cancer. JCI Insight 4, e129715 (2019).
Stelloo, S. et al. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol. Med. 7, 1450–1464 (2015).
Marcelli, M. et al. Quantifying effects of ligands on androgen receptor nuclear translocation, intranuclear dynamics, and solubility. J. Cell Biochem. 98, 770–788 (2006).
Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016).
Nadal, M. et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 8, 14388 (2017).
Sathya, G., Chang, C. Y., Kazmin, D., Cook, C. E. & McDonnell, D. P. Pharmacological uncoupling of androgen receptor-mediated prostate cancer cell proliferation and prostate-specific antigen secretion. Cancer Res. 63, 8029–8036 (2003).
McDonnell, D. P. et al. The mechanism of action of steroid hormones: a new twist to an old tale. J. Clin. Pharm. 33, 1165–1172 (1993).
Song, L. N., Coghlan, M. & Gelmann, E. P. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol. Endocrinol. 18, 70–85 (2004).
Sathya, G., Jansen, M. S., Nagel, S. C., Cook, C. E. & McDonnell, D. P. Identification and characterization of novel estrogen receptor-beta-sparing antiprogestins. Endocrinology 143, 3071–3082 (2002).
Xu, Y., Chen, S. Y., Ross, K. N. & Balk, S. P. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 66, 7783–7792 (2006).
Leung, J. K. & Sadar, M. D. Non-genomic actions of the androgen receptor in prostate cancer. Front Endocrinol. 8, 2 (2017).
Nguyen, T. V., Yao, M. & Pike, C. J. Androgens activate mitogen-activated protein kinase signaling: role in neuroprotection. J. Neurochem. 94, 1639–1651 (2005).
Unni, E. et al. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res. 64, 7156–7168 (2004).
Kang, H. Y. et al. Nongenomic androgen activation of phosphatidylinositol 3-kinase/Akt signaling pathway in MC3T3-E1 osteoblasts. J. Bone Min. Res. 19, 1181–1190 (2004).
Audet-Walsh, E. et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 31, 1228–1242 (2017).
Chen, Y., Han, L., Dufour, C. R., Alfonso, A. & Giguere, V. Canonical and nuclear mTOR specify distinct transcriptional programs in androgen-dependent prostate cancer cells. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.MCR-23-0087 (2023).
Geck, P., Maffini, M. V., Szelei, J., Sonnenschein, C. & Soto, A. M. Androgen-induced proliferative quiescence in prostate cancer cells: the role of AS3 as its mediator. Proc. Natl Acad. Sci. USA 97, 10185–10190 (2000).
McNair, C. et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Invest. 128, 341–358 (2018).
Gao, S. et al. Androgen receptor tumor suppressor function is mediated by recruitment of retinoblastoma protein. Cell Rep. 17, 966–976 (2016).
Zafarana, G. et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 118, 4053–4062 (2012).
Quigley, D. A. et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 175, 889 (2018).
Tran, C. et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 (2009).
Nyquist, M. D. et al. Supraphysiological androgens promote the tumor suppressive activity of the androgen receptor through cMYC repression and recruitment of the DREAM complex. Cancer Res. 83, 2938–2951 (2023).
Timmermans, S., Vandewalle, J. & Libert, C. Dimerization of the glucocorticoid receptor and its importance in (patho)physiology: a primer. Cells 11, 683 (2022).
Johnson, T. A., Paakinaho, V., Kim, S., Hager, G. L. & Presman, D. M. Genome-wide binding potential and regulatory activity of the glucocorticoid receptor’s monomeric and dimeric forms. Nat. Commun. 12, 1987 (2021).
Paakinaho, V., Johnson, T. A., Presman, D. M. & Hager, G. L. Glucocorticoid receptor quaternary structure drives chromatin occupancy and transcriptional outcome. Genome Res. 29, 1223–1234 (2019).
Caratti, B. et al. The glucocorticoid receptor associates with RAS complexes to inhibit cell proliferation and tumor growth. Sci. Signal 15, eabm4452 (2022).
Wade, H. E. et al. Multimodal regulation of E2F1 gene expression by progestins. Mol. Cell Biol. 30, 1866–1877 (2010).
Dolan, E. L. et al. Breast cancer cells can recognize and respond to different levels of progestins to achieve different phenotypic outputs. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/2023.10.13.562206v1 (2023).
Aggarwal, R. et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 36, 2492–2503 (2018).
Kotamarti, S., Armstrong, A. J., Polascik, T. J. & Moul, J. W. Molecular mechanisms of castrate-resistant prostate cancer. Urol. Clin. N. Am. 49, 615–626 (2022).
Fu, W. et al. Small-molecule inhibition of androgen receptor dimerization as a strategy against prostate cancer. ACS Cent. Sci. 9, 675–684 (2023).
El Kharraz, S. et al. N/C interactions are dispensable for normal in vivo functioning of the androgen receptor in male mice. Endocrinology 163, bqac104 (2022).
Sena, L. A. et al. Androgen receptor activity in prostate cancer dictates efficacy of bipolar androgen therapy through MYC. J. Clin. Invest. 132, e162396 (2022).
Grech, A., Breck, J. & Heidelbaugh, J. Adverse effects of testosterone replacement therapy: an update on the evidence and controversy. Ther. Adv. Drug Saf. 5, 190–200 (2014).
Achar, S., Rostamian, A. & Narayan, S. M. Cardiac and metabolic effects of anabolic-androgenic steroid abuse on lipids, blood pressure, left ventricular dimensions, and rhythm. Am. J. Cardiol. 106, 893–901 (2010).
Vigen, R. et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA 310, 1829–1836 (2013).
Basaria, S. et al. Adverse events associated with testosterone administration. N. Engl. J. Med 363, 109–122 (2010).
Cesarone, C. F., Bolognesi, C. & Santi, L. Improved microfluorometric DNA determination in biological material using 33258 Hoechst. Anal. Biochem. 100, 188–197 (1979).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Broad Institute, Picard Toolkit. Github Repository https://broadinstitute.github.io/picard/ (2019).
Team, R. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/060012v3 (2021).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21 29 21–21 29 29 (2015).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Stark, R. & Brown, G. DiffBind: differential binding analysis of ChIP-Seq peak data. Bioconductor http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2011).
Ross-Innes, C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Acknowledgements
We thank members of the McDonnell lab for their valuable input and discussion. This work was supported by NCI grant (1R01-CA271168 to DPM.) and NCI P30 Cancer Center Support Grant (CCSG, P30CA014236). This work used a high-performance computing facility partially supported by grants 2016-IDG-1013 (“HARDAC+: Reproducible HPC for Next-generation Genomics”) and 2020-IIG-2109 (“HARDAC-M: Enabling memory-intensive computation for genomics”) from the North Carolina Biotechnology Center.
Author information
Authors and Affiliations
Contributions
Conceptualization: RS, SEW, MB, SX, JDN and DPM. Methodology: RS, SEW, SP, TT, SN, MK. Investigation: RS, SEW, PW, SP, TK, AB, TT, SN, MK, MAN, MLK, EF, SWF, KO and DPM. Formal Analysis: XQ, ML, CF, ELD, TT, SN, MK, YX and HL. Resources: SR. Funding acquisition: DPM. Supervision: RS, SEW, KO, JDN and DPM. Writing – original draft: RS and DPM. Writing – review & editing: RS, XQ, ML, MB, SX, KO, JDN and DPM.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Safi, R., Wardell, S.E., Watkinson, P. et al. Androgen receptor monomers and dimers regulate opposing biological processes in prostate cancer cells. Nat Commun 15, 7675 (2024). https://doi.org/10.1038/s41467-024-52032-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52032-y
