
Abstract
Ibrutinib is a drug that inhibits the protein Burton’s tyrosine kinase and thereby the nuclear translocation of Nrf2, which played a key role in mediating the activation of antioxidants during stress conditions and ferroptosis resistance. This study aimed to identify the effect of Ibrutinib and ferroptosis inducer on colorectal cancer (CRC) treatment and its underlying mechanism. In our study, we found the upregulation of Nrf2 was correlated with CRC progression and antioxidant proteins. Ibrutinib sensitized CRC to ferroptosis inducers, suggested by further reduced CRC cell viability, proliferation and decreased antioxidant protein levels in CRC cells after combination treatment of Ibrutinib and RSL3 or Ibrutinib and Erastin both in vivo and in vitro. Knockout of Nrf2 diminished the regulatory effect of Ibrutinib on CRC sensitivity to ferroptosis inducers. Altogether, this study demonstrated that Ibrutinib increases the sensitivity of CRC cell to ferroptosis inducers by inhibiting Nrf2.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related death worldwide [1]. The majority of CRC cases were diagnosed in western countries; however, the incidence was increased in some countries due to its fast development [1]. Additionally, a high proportion of patients were detected at the advanced stages and had poor outcomes [2]. Therefore, it is urgent to discover new therapies for CRC.
Ferroptosis is a new mechanism of cancer cell death triggered by chemotherapeutic drugs [3]. Induction of ferroptosis results in mitochondrial dysfunction and toxic lipid peroxidation in cancer cells, which eventually induces the death of cancer cells [4]. CRC cell lines are insensitive to ferroptosis, but the underlying reasons remain to be elucidated [5]. Our preliminary data suggested that the resistance of CRC cell lines to ferroptosis may associate with Kelch-like ECH-associated protein 1(Keap1) deficiency, which was frequently found in CRC cells.
Keap1, a very cysteine-rich protein, is mainly found in the cytoplasm [6]. It is the principal negative regulator of nuclear factor erythroid 2 related factor 2 (Nrf2) by rapidly degrading Nrf2 [6]. The mutation or deficiency of Keap1 disrupts Keap1-Nrf2 interaction and results in overexpression of Nrf2 [7]. Nrf2, the key mediator of cellular oxidative response, was documented to be closely correlated with cancer cell ferroptosis resistance [8]. In oxidative stress conditions, Nrf2 protein regulates the transcriptional response of cells to oxidative stress and is translocated into the cell nucleus, which activates the antioxidants, including SOD1, CAT, and GSH-Px and reduces the accumulation of lipid peroxidation [9]. Keap1/Nrf2 expression levels were abnormally expressed in the majority of CRC patient tumor samples, such as reduced expression of Keap1, Keap1 mutation or increased accumulation of Nrf2 in cell nucleus in tumor tissues [10]. Based on this information, we speculated that the abnormal accumulation of Nrf2 in CRC nuclear may correlate with ferroptosis resistance of CRC cells. However, the role of Nrf2 nuclear translocation in ferroptosis resistance of CRC cells remains to be elucidated.
Bruton’s tyrosine kinase (BTK), a key component in the signal transduction pathway of B-cell antigen receptor (BCR), is considered a new potential target in cancer treatment [11]. Ibrutinib might irreversibly suppress BTK and BTK inhibition negatively mediated Nrf2 translocation into cell nucleus [11]. Thus, we hypothesized that Ibrutinib inhibited BTK, which prevented Nrf2 translocating to cell nucleus and the activation of the Nrf2 dependent antioxidant genes during oxidative stress conditions and eventually enhanced the sensitivity of CRC cells to ferroptosis.
Materials and Nethods
Animal model
Adult nude mice that were between 12–14 weeks and weighing 25–30 g were obtained from Guangdong Medical Laboratory Animal Center (Guangdong, China) and housed in an animal facility with a controlled environment. The animal experiments were approved by the Animal Ethical and Welfare Committee of Shenzhen University.
Sixty mice were randomly divided into six groups, (1) the CRC model group (model), (2) mice with RSL3 treatment, (3) mice with Erastin treatment, (4) mice with Ibrutinib treatment, (5) mice with RSL3 and Ibrutinib treatment, and (6) Erastin and Ibrutinib group. Murine subcutaneous tumor model and xenograft tumor mouse model were established and please refer to supplemental method for details.
For CRC model group, the mice were treated with PBS for two weeks. For RSL3 group, the mice were intraperitoneal injected with RSL3 (5 mg/kg daily) for two weeks. For Erastin group, the mice were intraperitoneal injected with Erastin (30 mg/kg, twice every other day) for two weeks. For Ibrutinib treatment group, mice were administered in drinking water at a concentration of 0.16 mg/ml for two weeks. Mice were also treated in combination with RSL and Ibrutinib or Erastin and Ibrutinib.
CRC tissue samples
CRC tissues were gathered from patients who were diagnosed at Shenzhen University General Hospital, and adjacent normal colorectal tissues collected were used as control. This study was approved by the Medical Ethics Committee of Shenzhen University, and informed consents were provided to each patient.
Histological analysis
Tumor tissue was fixed in 3.7% buffered formalin and was then embedded in paraffin. The samples were cut into 5 µm sections, which were stained with hematoxylin & eosin (H&E) using a previously described protocol [12]. The sections were imaged using a Biozero BZ-9000 Series microscope (KEYENCE, Osaka, Japan).
Immunohistochemistry staining
Mice tissues were fixed in 3.7% buffered formalin and were subsequently embedded in paraffin. Immunohistochemistry (IHC) staining was performed on 5 μm slides from embedded blocks. Slides were stained using the protocol as described previously [13]. The images were taken under a Leica confocal microscope.
Cell culture and treatment
NCI-H508, LoVo, LS513, HCT116, SW480, SW620, SW1116, DLD-1, HT-29, and Caco-2, CRC cell lines, were purchased from the American Type Culture Collection. Cells were maintained at DMEM supplemented with 10% FBS. Regarding the treatment, cells were seeded at 24 well-plates and were stimulated with serial concentrations of RSL3 (0, 0.1, 1, 5, or 10 μM) for 48 h or with Erastin (0, 2.5, 5, 10, 20, or 40 μM) for 48 h. The Caco-2 and HT-29 cell lines that were transfected with or without siNrf2 or correspondence siRNA were treated with RSL3 or Erastin together with Nrf2 inhibitor, NRF2-IN-1 (Merck) or Nrf2 activator, TAT-14 (Sigma-Aldrich) for 48 h. The Caco-2 and HT-29 cell lines that were transfected with or without siNrf2 or correspondence siRNA were treated with RSL3 or Erastin together with Ibrutinib (Imbruvica, 0.5 μM) for 48 h. Afterward, CRC cells were harvested for the following analyses.
Cell transfection
Based on previous studies, cell transfection was performed [13]. Cell lysates were harvested at 24 h after transfection.
Cell counting Kit-8 assay
CRC cell line proliferation was examined using a Cell counting Kit-8 (CCK-8) assay (Dojindo, Japan). The absorbance at 490 nm was obtained by a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Colony formation assay
Caco-2 and HT-29 cells were trypsinized and seeded in 6-well dishes with a density of 104 cells/well. After 24 h, cells were treated or transfected and treated based on the above-described method. 48 h later, the media was replaced with fresh media, followed by incubation of the well plates for 7 days. The number of colonies (>50 cells/colony) was counted under a Biozero BZ-9000 Series microscope.
Flow cytometry
Caco-2 and HT-29 were trypsinized and washed. The staining of cells with Annexin V and PI was performed utilizing a FITC Annexin V/PI Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) based on the manufacturer’s manual. The cells were analyzed using the FACS cytometer (BD Biosciences).
Lipid peroxidation assay
The relative malondialdehyde (MDA) concentration in cell lysates or in tissue extracts was detected using a Lipid Peroxidation Assay Kit (#ab118970, Abcam) based on the manufacturer’s protocol.
Dual-luciferase reporter assay
The assay was conducted according to a previously published protocol [14]. The Dual-Luciferase Reporter assay kit (Promega) was utilized to measure luciferase activities.
Western blot
Total cellular protein was extracted from the tumor tissues or the adjacent tissues or cells using the radio-immunoprecipitation assay buffer (Beyotime, Shanghai, China) with protease inhibitors for 30 min at 4 °C. For nuclear extracts, cells were lysed with 20 mM HEPES, which contained 10 mM NaCl, 1 mM DTT, 1.5 mM MgCl2, 0.2 mM EDTA, 20% glycerol and 0.1% NP-40. The samples were centrifuged and was lysed. Protein concentration was measured using a bicinchoninic acid protein assay kit (Pierce). Equal amounts of proteins (30 μg/well) were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), which were incubated with primary antibodies after blocking. The sections were incubated with secondary antibodies following visualization of the protein bands by an ECL kit (Millipore, Billerica, MA, USA).
Immunofluorescent staining
The cells were fixed with 4% PFA for about 20 min. The cells were blocked with the goat serum and then incubated with primary antibodies against Anti-Nrf2 (Abcam) at 4 °C overnight, followed by washing with PBS. After the incubation with the secondary antibody at room temperature for 1 h, the cells were incubated with DAPI. The images were taken under a confocal microscope (LSM710, Zeiss, Germany).
Statistical analysis
All statistical analysis was performed utilizing GraphPad Prism 5.0. All data are shown as mean ± standard deviation, which were from at least three independent experiments. An unpaired Student’s t test was utilized to compare two groups. The Pearson correlation analysis was performed to compare the correlation of Nrf2 expression with the SOD1, GPX4, and CAT. One-way analysis of variance (ANOVA) followed by the Turkey post hoc test was applied to compare multiple groups. P < 0.05 was considered significant.
Results
Increased NRF2 expression level is positively correlated to CRC progress
The Nrf2, SOD1, GPX4, and CAT expression levels are abnormally increased in the tissues from CRC patients compared to those in the corresponding paracancerous tissues (Fig. 1A, B). Significant positive correlations were detected between the expression levels of Nrf2 and that of SOD1, GPX4, and CAT according to the Pearson correlation test (Fig. 1C).

A The expressions of Nrf2, SOD1, GPX4, and CAT in tumor tissues and adjacent tissues from 10 CRC patients detected by western blot. B The expressions of Nrf2, SOD1, GPX4, and CAT in tumor tissues and adjacent tissues from 40 CRC patients detected by RT-qPCR. C The Pearson correlation analysis of Nrf2 with SOD1, CAT, and GPX4. All experiments were carried out at least three times, n = 3, *P < 0.05 and **P < 0.01.
In summary, increased Nrf2 expression level is positively correlated to CRC progression.
RSL3 and Erastin induces CRC ferroptosis
CRC cell lines were treated with a series of concentrations of Erastin or RSL3, the ferroptosis-inducing agents. Both RSL3 and Erastin significantly decreased CRC cell viability. Among these CRC cell lines, the Caco-2 and HT-29 cells showed the most and least reduced viability after RSL3 or Erastin treatment (Fig. 2A); therefore, these two cell lines were selected for the subsequent experiments. Compared to the controls, both RSL3 and Erastin reduced the colony formation in HT-29 and Caco-2 cells in a concentration-dependent manner (Fig. 2B). The impact of RSL3 or Erastin on Caco-2 cells was stronger than HT-29 cells, suggested that Caco-2 cells were more sensitive to the RSL3 or Erastin treatment than HT-29 cells. The content of end products of lipid peroxidation, including MDA, was significantly increased, but the GSH level was significantly decreased in HT-29 and Caco-2 cells after RSL3 or Erastin treatment in a concentration-dependent manner (Fig. 2C, D). Increased RSL3 or Erastin led to decreased antioxidants, SOD1, CAT, and GPX4 expression levels in HT-29 and Caco-2 cells (Fig. 2E). But RSL3 or Erastin treatments did not impact the expression of Nrf2 (Fig. 2E). Furthermore, the effect of RSL3 or Erastin treatment on MDA and GSH levels as well as the SOD1, CAT, and GPX4 expression levels in HT-29 cells was less than Caco-2 cells. Nuclear Nrf2 significantly increased luciferase activity of HT-29 cells and Caco-2 cells transfected with wt-SOD1, wt-CAT, or wt-GPX4 but not to those transfected with mut-SOD1, mut-CAT or mut-GPX4 (Fig. 2F). Treatment with RSL3 or Erastin had no significant impact of Nrf2 nuclear translocation to the luciferase activity of HT-29 cells transfected with wt-SOD1, wt-CAT, or wt-GPX4.

NCI-H508, LoVo, LS513, HCT116, SW480, SW620, SW1116, DLD-1, HT-29, and Caco-2, CRC cell lines, were stimulated with serial concentrations of RSL3 (0, 0.1, 1, 5, or 10 μM) for 48 h or were stimulated with Erastin (0, 2.5, 5, 10, 20, or 40 μM) for 48 h. The cell A viability and B proliferation were assessed via the CCK-8 assay and the colony formation assay. The C MDA, D GSH and E Nrf2, SOD1, CAT, and GPX4 levels were detected in HT-29 and Caco-2 cells that were treated with serial concentrations of RSL3 or Erastin. F The luciferase activity of HT-29 cells that were treated with or without Erastin or Caco-2 cells that treated with or without RSL3 were co-transfected with GPX4-WT, SOD1-WT or CAT-WT or GPX4-MUT, SOD1-MUT, or CAT-MUT and Nrf2. All experiments were carried out at least three times, n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001.
Altogether, RSL3 or Erastin treatment suppresses cell proliferation, reduces antioxidants and GSH level but increases MDA level in CRC cells. However, the treatment with RSL3 or Erastin has no impact on Nrf2 nuclear translocation.
Nrf2 inhibitor enhances the sensitivity of CRC cells to the ferroptosis induced by RSL3 or Erastin
To further investigate the role of Nrf2 in CRC cell resistance to ferroptosis, we treated the RSL3 and Erastin-induced CRC cells with Nrf2 inhibitor, NRF2-IN-1, or Nrf2 activator, TAT-14. As the HT-29 cells showed less sensitivity to the RSL3 or Erastin treatment compared to the Caco-2 cells, the HT-29 cells were chosen for further experiments to represent the worst scenario. The treatment of NRF2-IN-1, together with RSL3 or Erastin resulted in a synergistic inhibitory effect on the HT-29 viability and colony formation (Fig. 3A, B). The additional treatment with NRF2-IN-1 led to a stronger increasing of MDA level and a stronger reduction of GSH level in HT-29 cells in comparison to the treatment of RSL3 or Erastin alone (Fig. 3C, D). The NRF2-IN-1 treatment significantly reduced Nrf2 expression as indicated by IF figures and western blot results (Fig. 3E, F); in addition, the treatment of RSL3 or Erastin augmented the inhibitory effect of NRF2-IN-1 on Nrf2 expression. The combination treatments of NRF2-IN-1 and RSL3 or NRF2-IN-1 and Erastin also had a synergistic inhibitory effect on the SOD1, CAT and GPX4 expression levels (Fig. 3F).

HT-29 cells were treated with RSL3 or Erastin with or without the presence of Nrf2 inhibitor, NRF2-IN-1. The cell (A) viability and (B) proliferation were assessed via the CCK-8 assay and the colony formation assay. The C MDA and D GSH levels were detected in HT-29 cells that were treated with RSL3 or Erastin with or without the presence of Nrf2 inhibitor, NRF2-IN-1. E The representative immunofluorescent staining images of Nrf2 in HT-29 cells that were treated with RSL3 or Erastin with or without the presence of Nrf2 inhibitor, NRF2-IN-1. Scale bar = 25 μm. F The Nrf2, GPX4, SOD1, and CAT expressions in HT-29 cells that were treated with RSL3 or Erastin with or without the presence of Nrf2 inhibitor, NRF2-IN-1. Caco-2 cells were treated with RSL3 or Erastin with or without the presence of Nrf2 promotor, TAT-14. The cell G viability and H proliferation were assessed via the CCK-8 assay and the colony formation assay. The I MDA and J GSH levels were detected in Caco-2 cells that were treated with RSL3 or Erastin with or without the presence of Nrf2 promotor, TAT-14. K The representative immunofluorescent staining images of Nrf2 in Caco-2 cells that were treated with RSL3 or Erastin with or without the presence of Nrf2 promotor, TAT-14. Scale bar = 25 μm. L The Nrf2, GPX4, SOD1, and CAT expressions in Caco-2 cells that were treated with RSL3 or Erastin with or without the presence of Nrf2 promotor, TAT-14. All experiments were carried out at least three times, n = 3, *P < 0.05, **P < 0.01, and ***P < 0.001.
TAT-14, the activator of Nrf2, attenuated the inhibitory effect of RSL3 or Erastin treatment on the Caco-2 viability and colony formation (Fig. 3G, H). Furthermore, the treatment of TAT-14 suppressed the stimulatory effect of RSL3 or Erastin treatment on the MDA level (Fig. 3I). RSL3 or Erastin treatment significantly reduced GSH level in Caco-2 cells, but TAT-14 showed an opposite effect (Fig. 3J). Similarly, the Nrf2 nuclear translocation was induced in TAT-14-treated Caco-2 cells as indicated by immunofluorescent staining and western blot data (Figs. 3K, L and S1A). However, the induction of Nrf2 nuclear translocation was attenuated by RSL3 or Erastin treatment. TAT-14 treatment also increased SOD1 and CAT expression levels as indicated in Fig. 3L. Additionally, RSL3 or Erastin-induced reduction of SOD1, CAT and GPX4 levels was restored by TAT-14 treatment.
Collectively, these data suggest that suppression of Nrf2 enhances CRC cell sensitivity to RSL3 or Erastin-induced ferroptosis but treatment with Nrf2 activator has an opposite effect.
Ibrutinib sensitizes CRC cells to the ferroptosis induced by RSL3 or Erastin
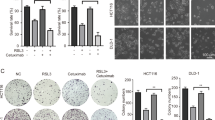
The combination treatments of Ibrutinib and RSL3 or Ibrutinib and Erastin showed a synergistic effect inhibiting the HT-29 and Caco-2 cells viability and colony formation (Fig. 4A, B). The combined treatment with Ibrutinib and RSL3 or Ibrutinib and Erastin further increased MDA level but reduced GSH level in HT-29 and Caco-2 cells compared to those only treated with RSL3 or Erastin (Fig. 4C, D). Ibrutinib treatment decreased nuclear translocation of Nrf2 in HT-29 and Caco-2 cells and additional treatment with RSL3 or Erastin further reduced Nrf2 nuclear translocation (Fig. 4E, F and S1B). Similarly, Ibrutinib inhibited the expression levels of GPX4, SOD1 and CAT (Fig. 4F). The combination treatments of Ibrutinib and RSL3 or Erastin showed a synergistic inhibition effect on GPX4, SOD1, and CAT expression levels.

A The viability and B proliferation of HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3 or Ibrutinib/Erastin detected by the CCK-8 assay and the colony formation assay. The C MDA and D GSH levels in HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3, or Ibrutinib/Erastin detected by the commercial kits. E The representative immunofluorescent staining images of Nrf2 in HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3, or Ibrutinib/Erastin. Scale bar = 25 μm. F The Nrf2, GPX4, SOD1, and CAT expressions in HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3, or Ibrutinib/Erastin detected by Western blot. All experiments were carried out at least three times, n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001.
Taken together, Ibrutinib treatment may increase the sensitivity of CRC cells to ferroptosis.
Nrf2 plays a critical role in Ibrutinib regulates the sensitivity of CRC cells to RSL3 or Erastin-induced ferroptosis
To further identify the role of Nrf2 in Ibrutinib regulated the sensitivity of CRC cells to RSL3 or Erastin induction, we knocked out Nrf2 in HT-29 cells by specific shRNA (Nrf2-KO group). After knockout of Nrf2 in HT-29 cells, Ibrutinib treatment did not show any impact on HT-29 cell viability and colony formation (Fig. 5A, B). The synergistic inhibitory effect of combination treatments of Ibrutinib and RSL3 or Erastin on HT-29 cell viability and colony formation was disappeared as well after knockout of Nrf2 (Fig. 5A, B). In addition, Ibrutinib further increased MDA level and decreased GSH level in Nrf2-wildtype HT-29 cells in the absence or presence of RSL3 or Erastin but not in the Nrf2-knockout group (Fig. 5C, D). As expected, the Nrf2 expression could not be detected in the absence or presence of Ibrutinib in the Nrf2-knockout group (Fig. 5E). Ibrutinib treatment had no impact on GPX4, SOD1, and CAT expression levels in HT-29 cells following RSL3 or Erastin treatment after knockout of Nrf2. Altogether, these data indicate that Nrf2 plays a key role in the Ibrutinib-mediated sensitivity of CRC cells to RSL3 or Erastin-induced ferroptosis.

The impact of Nrf2 knockout on the A viability and B proliferation of HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3, Ibrutinib/Erastin, NRF2-IN-1/RSL3, or Erastin/NRF2-IN-1 detected by the CCK-8 assay and the colony formation assay. The impact of Nrf2 knockout on the C MDA and D GSH levels in the HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3, Ibrutinib/Erastin, NRF2-IN-1/RSL3, or Erastin/NRF2-IN-1. E The impact of Nrf2 knockout on Nrf2, GPX4, SOD1 and CAT expressions in HT-29 and Caco-2 cells that were treated with Ibrutinib, RSL3, Erastin, Ibrutinib/RSL3, Ibrutinib/Erastin, NRF2-IN-1/RSL3, or Erastin/NRF2-IN-1. All experiments were carried out at least three times, n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001.
Ibrutinib sensitizes CRC tumors to RSL3 or Erastin-induced ferroptosis in vivo
To determine whether Ibrutinib enhances the sensitivity of CRC tumors to RSL3 or Erastin-induced ferroptosis in vivo, the HT-29 cells were subcutaneously implanted to the nude mice. After 7 days, these mice were treated with RSL3, Erastin, Ibrutinib, RSL3 and Ibrutinib, or Erastin and Ibrutinib. Compared to the control group, treatments with RSL3, Erastin and Ibrutinib effectively reduced the tumor sizes (Fig. 6A, B) with decreased expression levels of GPX4, SOD1 and CAT (Fig. 6C). Combination treatments of Ibrutinib and RSL3 or Ibrutinib and Erastin further decreased tumor sizes in relation to the control group and the single treatment group with further reduced expression levels of Nrf2, GPX4, SOD1 and CAT. We also investigated the presence of lung tumors because it is the common site for metastases in human CRC [15]. The CRC suspension that derived from CRC tumors from the patients was intravenously injected through the tail vein into mice and after seven days, the mice were divided into six groups: control, RSL3, Erastin, Ibrutinib, RSL3 and Ibrutinib, or Erastin and Ibrutinib. The treatment of RSL3 and Ibrutinib or Erastin and Ibrutinib significantly reduced the lung injury and the number of lung metastasis as indicated by H&E staining and its quantification result (Fig. 6D–F). The treatment of Ibrutinib combined RSL3 or Erastin had a synergistic effect. A single treatment of Ibrutinib decreased the expression of Nrf2, SOD1 and CAT (Fig. 6G). The treatment of RSL3 or Erastin alone did not have a significant impact on Nrf2, expression levels but reduced SOD1, CAT, and GPX4 expression. The Nrf2, SOD1, CAT and GPX4 expression levels were obviously decreased in CRC tumors of mice that were treated with both RSL3 and Ibrutinib or Erastin and Ibrutinib.

The HT-29 cells were implanted into the subcutaneous space of the nude mice. After seven days, these mice were treated with RSL3, Erastin, Ibrutinib, RSL3 and Ibrutinib, or Erastin and Ibrutinib. A The images and B its quantification results of CRC tumors from different groups. C The expressions of Nrf2, GPX4, SOD1, and CAT in mice that were treated with RSL3, Erastin, Ibrutinib, RSL3 and Ibrutinib, or Erastin and Ibrutinib. D–F The CRC suspension that derived from CRC tumors from the patients was intravenously injected through the tail vein into mice and after seven days, the mice were RSL3, Erastin, Ibrutinib, RSL3 and Ibrutinib, or Erastin and Ibrutinib. The images of lung, the quantification results of lung metastasis numbers calculated from lung sections and the H&E staining of lung sections from different groups. Scale bar = 100 μm. G The representative immunohistochemistry images of lung sections from different groups indicate the Nrf2, GPX4, SOD1, and CAT expressions. Scale bar = 100 μm. All experiments were carried out at least three times, n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001.
Taken together, Ibrutinib increases the sensitivity of CRC mice to RSL3 or Erastin-induced ferroptosis.
Discussion
Ferroptosis is characterized by inhibition of the GPX4 and accumulation of intracellular lipid reactive oxygen species [16]. It can be activated by RSL3 and Erastian. RSL3 negatively targets GPX4, which regulates the suppression of ferroptosis [17]. Erastin may induce ferroptosis by inhibiting the cysteine-glutamate antiporter, which leads to the inactivation of the cellular antioxidant GSH [17]. Interestingly, both GPX4 and GSH are the downstream targets of Nrf2 [3]. In this study, we demonstrated the correlation of Nrf2 nuclear translocation in CRC progression and increased antioxidants. The treatment of Ibrutinib can prevent Nrf2 translocating to nucleus, which sensitizes the CRC to ferroptosis inducing agents, RSL3 and Erastin, both in vitro and in vivo. This may provide a novel theoretical basis for the application of Ibrutinib and ferroptosis inducers in CRC treatment.
It was documented previously that the Nrf2 has a complex and dual role in CRC [18]. The activation of the Nrf2 pathway may protect cancer cells in an early stage of tumorigenesis but prevent tumor progression and cancer metastasis at a later stage [18]. However, the role of CRC remains to be investigated. In this study, we detected that the Nrf2 translocation into nucleus was increased in the CRC tumor compared to the normal tissue, suggesting that the nuclear translocation of Nrf2 was correlated with the progression stage of CRC. Consistently with our study, a recent publication indicated that the expression of Nrf2 was independent of patients’ sex, age, invasion and location, but significantly correlated with stage classification [19]. During stress conditions, the Nrf2 is disassociated from its repressor Keap1 and translocated to the nucleus, which thereby mediated cellular stress response genes by its transcriptional activity [20]. In this study, we further demonstrated that the upregulation of Nrf2 in advanced stage CRC was associated with increased antioxidant proteins, including GPX4, SOD1 and CAT, which may reduce lipid peroxidation and thus inhibited ferroptosis. The RSL3 or Erastin treatment of CRC cell lines did not change the nuclear translocation of Nrf2 in CRC cell lines but significantly reduced antioxidant proteins, which suggest that the RSL3 or Erastin treatment did not impact the activation of Nrf2 signaling but induced ferroptosis by suppression antioxidant proteins. The reduced level of MDA in the RSL3 or Erastin treated CRC cell lines further confirmed this.
Previous studies demonstrated that suppression of Nrf2 can significantly increase the sensitivity of several cancer cells, including carcinoma cells and glioma cells, to pro-ferroptosis agents [21]. Sun et al. [22] showed that knockdown of Nrf2 in hepatocellular carcinoma cells enhanced the treatment efficacy of Erastin and Sorafenib both in vitro and in vivo. Another study documented that the Nrf2-Keap1 signaling pathway played a critical role in regulating glioma cell resistance to cell death processes, including ferroptosis [23]. Furthermore, Nrf2 inhibitor has been used to increase efficacy of chemotherapeutic agents in several cancer types [24]. It was shown that inhibition of Nrf2 with Nrf2 inhibitor strongly increased the reaction of three cancer cell lines, neuroblastoma, breast adenocarcinoma and non-small cell lung carcinoma to anticancer drugs [24]. Consistently, our data demonstrated that the combination treatment of Nrf2 inhibitor or Ibrutinib with RSL3 or Erastin had a synergistic effect on inhibition of CRC cell proliferation, viability as well as antioxidant proteins. After knockout of Nrf2, the synergistic inhibitory effect of Ibrutinib and RSL3 or Erastin on CRC cell viability, proliferation and antioxidant proteins in CRC cell lines were diminished. These data indicated that the treatment of Nrf2 inhibitor or Ibrutinib sensitized the RSL3 or Erastin-induced ferroptosis by suppressing Nrf2.
Finally, we confirmed the in vitro data using the established mouse CRC model. The combination treatments of Ibrutinib and RSL3 or Erastin had a synergistic effect on suppressing the CRC tumors in terms of tumor sizes and the antioxidant proteins in the tumors, suggesting Ibrutinib sensitized CRC tumors to the RSL3 or Erastin induced ferroptosis. In addition, the Ibrutinib and RSL3 or Irbutinib and Erastin further reduced lung metastasis, Nrf2 and antioxidant proteins in CRC mouse models compared to the single treatment and non-treated controls.
In conclusion, this study reported that Ibrutitib sensitizes CRC to RSL3 or Erastin-induced ferroptosis by inhibiting Nrf2. Our study showed that the combination treatment of Nrf2 inhibition agents with ferroptosis inductors may be a potential strategy for CRC treatment.
Data availability
All data generated or analyzed during this study are included in this article. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Change history
07 February 2024
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41419-024-06498-9
References
Yang Y, Han Z, Li X, Huang A, Shi J, Gu J. Epidemiology and risk factors of colorectal cancer in China. Chin J Cancer Res. 2020;32:729–41.
Bray F, Ferlay J, Soerjomataram I, Siegel R, Torre L, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107.
Wu C, Zhao W, Yu J, Li S, Lin L, Chen X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci Rep. 2018;8:574.
Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci. 2021;17:2703–17.
Sadeghi MR, Jeddi F, Soozangar N, Somi MH, Samadi N. The role of Nrf2-Keap1 axis in colorectal cancer, progression, and chemoresistance. Tumour Biol. 2017;39:1010428317705510.
Kopacz A, Kloska D, Forman HJ, Jozkowicz A, Grochot-Przeczek A. Beyond repression of Nrf2: an update on Keap1. Free Radic Biol Med. 2020;157:63–74.
Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267.
Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer. 2020;122:279–92.
Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. 2016;15:71.
Lee Y, Kim W, Bae J, Cho M, Lee S, Nam H, et al. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann Surg Treat Res. 2020;98:159–67.
Cabiati M, Sapio A, Salvadori C, Burchielli S, Carlucci L, Mattii L, et al. Evaluation of transcriptional levels of the natriuretic peptides, endothelin-1, adrenomedullin, their receptors and long non-coding RNAs in rat cardiac tissue as cardiovascular biomarkers of aging. Peptides. 2020;123:170173.
Koukourakis MI, Giatromanolaki A, Sivridis E, Simopoulos C, Gatter KC, Harris AL, et al. LYVE-1 immunohistochemical assessment of lymphangiogenesis in endometrial and lung cancer. J Clin Pathol. 2005;58:202–6.
Chen X, Sun R, Yang D, Jiang C, Liu Q. LINC00167 regulates RPE differentiation by rargeting the miR-203a-3p/SOCS3 Axis. Mol Ther Nucleic Acids. 2020;19:1015–26.
Riihimäki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:29765.
Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267.
Li J, Cao F, Yin H-L, Huang Z-J, Lin Z-T, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88.
Gonzalez-Donquiles C, Alonso-Molero J, Fernandez-Villa T, Vilorio-Marqués L, Molina AJ, Martín V. The NRF2 transcription factor plays a dual role in colorectal cancer: A systematic review. PLoS ONE. 2017;12:e0177549.
Lee YJ, Kim WI, Bae JH, Cho MK, Lee SH, Nam HS, et al. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann Surg Treat Res. 2020;98:159–67.
Deshmukh P, Unni S, Krishnappa G, Padmanabhan B. The Keap1-Nrf2 pathway: promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys Rev. 2017;9:41–56.
Yan H, Zou T, Tuo Q, Xu S, Li H, Belaidi A, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49.
Wang X, Sun Z, Villeneuve N, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–43.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Fan Z, Wirth A, Chen D, Wruck C, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371.
Acknowledgements
This work was supported by Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515012613) & Health Science and Technology Program of Nanshan District (No. NS2021156) & Natural Science Foundation of Shenzhen University General Hospital (No. SUGH2020QD024) & Sanming Project of Medicine in Shenzhen (No. SZSM202111004) & Shenzhen Key Medical Discipline Construction Fund.
Author information
Authors and Affiliations
Contributions
JFZ: conceptualization, writing-original draft, methodology, and formal analysis; YL: supervision; WTL: validation; MHL: data curation; CHZ: resources; PWS: investigation; JXC: software; WHW: visualization; WZ: funding acquisition, project administration, and writing-review & editing. All authors have read and approved the final version of this manuscript to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The animal experiments were approved by the Animal Ethical and Welfare Committee of Shenzhen University. This study was approved by the Medical Ethics Committee of Shenzhen University, and informed consents were provided to each patient.
Informed consent
The informed consent was obtained from study participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Andreas Linkermann
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41419-024-06498-9"
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, JF., Liu, Y., Li, WT. et al. RETRACTED ARTICLE: Ibrutinib facilitates the sensitivity of colorectal cancer cells to ferroptosis through BTK/NRF2 pathway. Cell Death Dis 14, 151 (2023). https://doi.org/10.1038/s41419-023-05664-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-05664-9
This article is cited by
-
The mechanism of ferroptosis and its related diseases
Molecular Biomedicine (2023)
-
Advancing the frontiers of colorectal cancer treatment: harnessing ferroptosis regulation
Apoptosis (2023)
