
Abstract
Mutations in mitochondrial energy-producing genes lead to a heterogeneous group of untreatable disorders known as primary mitochondrial diseases (MD). Leigh syndrome (LS) is the most common pediatric MD and is characterized by progressive neuromuscular affectation and premature death. Here, we show that daily cannabidiol (CBD) administration significantly extends lifespan and ameliorates pathology in two LS mouse models, and improves cellular function in fibroblasts from LS patients. CBD delays motor decline and neurodegenerative signs, improves social deficits and breathing abnormalities, decreases thermally induced seizures, and improves neuropathology in affected brain regions. Mechanistically, we identify peroxisome proliferator-activated receptor gamma (PPARγ) as a key nuclear receptor mediating CBD’s beneficial effects, while also providing proof of dysregulated PPARγ expression and activity as a common feature in both mouse neurons and fibroblasts from LS patients. Taken together, our results provide the first evidence for CBD as a potential treatment for LS.
Similar content being viewed by others


Introduction
Mitochondria generate the majority of cellular ATP1. Mutations in either nuclear or mitochondrial genomes causing mitochondrial dysfunction lead to a diverse group of disorders known as primary mitochondrial diseases (MDs), affecting predominantly high energy-requiring organs/tissues2,3. MDs are usually progressive, causing significant disability and, in many cases, premature death. Among them, Leigh syndrome (LS), or subacute necrotizing encephalopathy, is the most common form of pediatric MD. LS is characterized by neuromuscular affectation with symmetrical lesions in the basal ganglia and brainstem4,5,6.
Mutations in >75 genes encoding mitochondrial proteins, such as the complex I subunit NDUFS4, have been described to cause LS7. Mice lacking Ndufs4 (Ndufs4KO) recapitulate several LS features, including difficulty to thrive, motor problems, breathing abnormalities, seizures, significantly shortened lifespan, neuroinflammation, and neurodegeneration8,9,10. Overall, the prognosis of LS and other MDs is very poor. No cure exists at present and patients rely on palliative treatments with limited effectivity11,12,13. This emphasizes the need for novel, effective treatments to alleviate suffering and/or increase survival in MD patients.
Accumulating evidence suggests that cannabis-derived compounds show therapeutic value to some neurodegenerative disorders, which share common features with MDs such as oxidative stress, neuroinflammation, and neuronal death14,15. Moreover, cannabinoids have also been reported to exert beneficial effects in some cases of epilepsy and pain, two common traits observed in several MDs16, including LS. Among the myriad of existing cannabinoids, cannabidiol (CBD) has been gaining attention for its broad therapeutic properties and its low side-effect profile. CBD possesses anti-inflammatory, antioxidant, neuroprotective and anticonvulsant properties15,17,18,19,20, which are of high interest in the context of LS. Indeed, CBD received FDA approval for the treatment of seizures associated with three other pediatric syndromes21. However, whether CBD also exerts antiseizure activity in mitochondrial epilepsy or whether CBD may improve survival and comorbidities associated with LS remains to be elucidated.
Here we report that daily CBD administration significantly extends lifespan and improves clinical signs in mouse models of two distinct LS phenotypic presentations, identifying downstream targets for the beneficial effects of CBD and paving the way for novel therapeutic avenues for LS.
Results
CBD prolongs lifespan and improves fitness in Ndufs4-deficient mice
Ndufs4 knockout (Ndufs4KO) mice, a well characterized mouse model of LS, display a progressive neurodegenerative phenotype, which resembles the human disease including retarded growth rate, lethargy, loss of motor skills, and premature death8,9. To assess the therapeutic potential of CBD, we first examined the effects of daily CBD treatment (100 mg/kg, i.p.) beginning promptly after weaning (Postnatal day [PND] 23-29). CBD treatment significantly extended the lifespan of Ndufs4KO mice. Median survival was 57 and 70 days for vehicle and CBD groups, respectively (Fig. 1a). CBD treatment significantly delayed the onset of common clinical signs associated with neurological decline, such as clasping, twisting, and curling22, compared to vehicle-treated mice (Fig. 1b). A progressive decrease in motor function also appears as a consequence of the neurological decline present in Ndufs4KO mice22. CBD treatment also delayed this observed motor decline (Fig. 1c), as measured by rotarod tests conducted at PND30, 40 and 50.

a Kaplan–Meier survival curve of Ndufs4KO mice treated with vehicle (n = 26, gray) and CBD (n = 25, red, Log-rank test ****p < 0.0001), and control mice (n = 20, black). b Clasping/twisting onset in Ndufs4KO treated with vehicle (n = 14, gray) and CBD (n = 11, red, unpaired two-tailed t-test, ****p < 0.0001). c Rotarod performance in vehicle- and CBD-treated Ndufs4KO (n = 19 mice/group) and control mice (n = 15 mice/group, three-way ANOVA followed by Tukey’s multiple comparisons test, **p = 0.0076, ***p = 0.0003, ****p < 0.0001 [genotype effect]; #p = 0.0278, ###p = 0.0004 [treatment effect]). Time to fall off is represented among the 3 tested days (PND30, 40, and 50). Respiratory tidal volume (d), frequency (e), and total ventilation (f) of vehicle- (n = 6 mice) and CBD-treated Ndufs4KO (n = 7 mice) and control mice (n = 11 mice/group, two-way ANOVA followed by Tukey’s multiple comparisons test, **p < 0.01, ***p < 0.001, ****p < 0.0001 [genotype effect]; #p < 0.05, ##p < 0.01, ####p < 0.0001 [treatment effect]). Boxplots show the median (horizontal line), upper and bottom quartiles (box outlines) and the maximum and minimum non-outlier data points (whiskers). All data are presented as mean values ± SEM. All source data are provided as a source data file. Veh vehicle, CT control mice, KO Ndufs4KO mice, VT Tidal volume, fR Respiratory frequency, VE Total ventilation.
Respiratory arrest is the most common cause of death for LS patients. Accordingly, Ndufs4KO mice present similar breathing abnormalities9, which may be a response to metabolic acidosis10. These mice have normal respiratory frequency (fR) but display a higher tidal volume (VT) than control mice9 (Fig. 1d, e). Accordingly, their product, i.e. total ventilation or minute ventilation (VE = fR · VT), is increased in Ndufs4KO mice9 (Fig. 1f). We found that daily CBD treatment normalized the altered total ventilation (Fig. 1f) by decreasing respiratory frequency (Fig. 1e), without direct effect on tidal volume (Fig. 1d). These results show that chronic CBD administration delays both the neurological and motor decline, improves breathing abnormalities, and ultimately improves survival in Ndufs4KO mice.
CBD extends lifespan and reduces seizures in a MD-mediated fatal epilepsy mouse model
The severity of the Ndufs4KO phenotype hinders comprehensive characterization of the underlying pathophysiological mechanisms. Thus, we took advantage of conditional knockout mice that lack Ndufs4 only in GABAergic cells (Gad2:Ndufs4cKO mice)23 to dissect specific clinical manifestations that are common in MD children such as epilepsy and autistic behaviors24. Gad2:Ndufs4cKO mice show no signs of motor impairments but present recurrent seizures that lead to death by ~PND7023 (Fig. 2a), thus presenting phenotypic distinction from Ndusf4KO and an adequate model of MD-related epilepsy. While the exact mechanisms underlying this model’s particular sensitivity to fatal seizures remain unknown, GABAergic interneuron loss, imbalanced excitatory and inhibitory input as well as insufficient metabolic capacity to recover from seizures may be involved25. Given the short half-life of CBD in rodents (~4.5 h)26 compared to humans (~24 h)27, we administered CBD (100 mg/kg, i.p.) twice daily beginning before the period of seizure risk (PND33-37) to ensure an effective CBD concentration against spontaneous seizures28. Repeated CBD treatment robustly extended the lifespan of Gad2:Ndufs4cKO mice (median survival: 64 vs. 100 days in vehicle- and CBD-treated Gad2:Ndufs4cKO mice respectively; Fig. 2a) in a dose-dependent manner, with 100 mg/kg being the most effective dose, followed by 200 mg/kg (Supplementary Fig. S1a). No effect was detected at lower doses (25 mg/kg and 50 mg/kg). Administration of a rapamycin dose regimen that has previously been shown to rescue Ndufs4KO lifespan (8 mg/kg daily, i.p.) did not increase lifespan in Gad2:Ndufs4cKO mice when treatment was initiated at the same stage as CBD (Supplementary Fig. S1b, c).

a Kaplan–Meier survival curve of Gad2:Ndufs4cKO mice treated daily with vehicle (n = 24, gray) and CBD (n = 18, red, Log-rank test ****p < 0.0001), and control mice (n = 20, black). b % of Gad2:Ndufs4cKO mice acutely treated with vehicle (n = 5) or CBD (100 mg/kg, n = 7) upon thermally induced seizure onset that survived for the next 8 h. c % of Gad2:Ndufs4cKO mice acutely treated with vehicle (n = 5) or CBD (100 mg/kg, n = 5) 1 h before thermally inducing seizures that displayed (gray) or not (white) tonic-clonic seizures at some point during the next 4 h after seizure onset. Discrimination index of the time exploring a stranger mouse over an empty wire cage (d) and total exploration time (e) in the 3-chamber social interaction test of Gad2:Ndufs4cKO and control mice treated daily with vehicle or CBD (n = 12 [control-vehicle and Gad2:Ndufs4cKO-CBD], 16 [control-CBD and Gad2:Ndufs4cKO-vehicle], two-way ANOVA followed by Tukey’s multiple comparisons test, ****p < 0.0001 [genotype effect]; ####p < 0.0001 [treatment effect]). % of time spent in open arms (f), % of entries in open arms (g), and total arm entries (h) in the elevated plus maze test of Gad2:Ndufs4cKO and control mice treated daily with vehicle or CBD (n = 15 [control-vehicle], 22 [Gad2:Ndufs4cKO-vehicle], 13 [control-CBD], 11 [Gad2:Ndufs4cKO-CBD], two-way ANOVA followed by Tukey’s multiple comparisons test, ****p < 0.0001 [genotype effect]; ##p = 0.0055, ####p < 0.0001 [treatment effect]). All data are presented as mean values ± SEM. All source data are provided as a source data file. Veh vehicle, CT control mice, cKO Gad2:Ndufs4cKO mice.
Drug-resistant epilepsy is a common neurologic manifestation of LS29, which is frequently observed after stressors such as febrile episodes in LS patients30. Accordingly, Gad2:Ndufs4cKO mice are susceptible to thermally induced seizures, and the number and duration of seizures significantly increase with temperature23. To test the antiepileptic effect of CBD administration, a cohort of Gad2:Ndufs4cKO mice received CBD (100 mg/kg) or vehicle coincidently with the onset of thermally induced seizures. CBD treatment led to an increased survival rate in Gad2:Ndufs4cKO (40% of vehicle-treated vs. 71% CBD-treated, Supplementary Fig. S2a). To assess whether CBD pre-treatment could prevent the appearance of epileptic events, a second cohort of Gad2:Ndufs4cKO mice were treated with CBD (100 mg/kg) 1 h prior to the thermal-induction procedure. A 100% survival rate was observed in CBD-pretreated Gad2:Ndufs4cKO mice (Fig. 2b), coincident with a lower presence of generalized tonic-clonic seizures after thermal induction compared to vehicle (Fig. 2c). All control mice survived without seizures (Fig. 2b, c).
Lack of interneuron function can lead to cortical hyperexcitability, altering brain activity profiles. In this regard, hyperexcitability has been associated to cognitive impairment and epileptic activity31. Thus, we measured cortical neural activity by EEG in Gad2:Ndufs4cKO mice that received either vehicle or CBD immediately after the onset of a thermally induced seizure. Vehicle-treated Gad2:Ndufs4cKO mice displayed abnormal EEG features, which were found to be reduced in both intensity and duration in CBD-treated mice (Supplementary Fig. S2b). Collectively, these results suggest that CBD can both prevent and reduce MD-related seizures.
To further dissect the potential contribution of CBD against seizure progression, we recorded extracellular field excitatory postsynaptic potential (fEPSP) from the hippocampal CA1 region of Gad2:Ndufs4cKO mice before and after bath application of picrotoxin (100 µM, 8–10 min), to estimate the inhibitory/excitatory ratio of CA3-CA1 synapses, a relevant circuit for memory and epilepsy32. As expected, control animals showed a significant increase in fEPSP after picrotoxin application, which was missing in mutant animals, indicating a defect in inhibitory transmission (Supplementary Fig. S2c). In agreement with previous reports on CBD action onto hippocampal synapses28,33,34, a CBD pre-treatment (16 µM, 1 h treatment prior recordings) blunted picrotoxin-induced excitatory transmission in both experimental groups, although excitatory transmission restoration to basal levels appeared subtler in mutant mice than in control subjects given their reduced inhibitory tone (Supplementary Fig. S2c). Altogether, these data indicate that CBD may play a restorative effect counteracting epileptogenic activity due to the loss of inhibitory transmission.
CBD improves behavioral comorbidities in a MD-mediated fatal epilepsy mouse model
Autism-spectrum traits are increasingly being recognized as prevalent manifestations in primary MDs24. Accordingly, Gad2:Ndufs4cKO mice display significant social deficits compared to control littermates in the three-chamber social approach test (Fig. 2d), validating their use as a genetic model for MD-related autistic-like behavior. To test CBD’s efficacy in treating decreased sociability, we performed the three-chamber social approach test in Gad2:Ndufs4cKO and control mice repeatedly treated with CBD (100 mg/kg) or vehicle 1 h before testing. CBD treatment reversed the autistic-like behavioral traits observed in Gad2:Ndufs4cKO mice, as shown by an increased time spent exploring the stranger mouse compared to the object (Fig. 2d). No alterations in total exploration time were observed (Fig. 2e). Doses of 50 and 200 mg/kg also reversed social deficits in Gad2:Ndufs4cKO mice, whereas 25 mg/kg dose had no effect (Supplementary Fig. S1d). No differences in total exploration times were observed between treatments (Supplementary Fig. S1e).
In addition to social deficits, Gad2:Ndufs4cKO mice presented an increased percentage of time spent and entries in the open arms (Fig. 2f, g) in the elevated plus maze test, suggesting either an altered anxiety-like behavior or impaired threat perception. No differences in total arm entries were observed between groups (Fig. 2h). Again, CBD treatment rescued this phenotype in this test in a dose-dependent manner (Supplementary Fig. S1f–h). No behavioral effects of CBD (100 mg/kg) were observed in control mice. Overall, daily CBD administration prolongs survival, reduces seizures, rescues social deficits and anxiety-related behaviors in Gad2:Ndufs4cKO mice.
CBD attenuates neuroinflammation in a LS mouse model
Neurological symptoms in LS mouse models strongly correlate with neuroinflammation35,36. In this regard, Gad2:Ndufs4cKO mice display gliosis –including both microglial (IBA1+ cells) and astroglial (GFAP+ cells) reactivity– in discrete brain areas, such as basal ganglia23, recapitulating the lesions found in LS patients4,5,6. Given the broad anti-inflammatory effects attributed to CBD17,37, we set to assess whether CBD also affects neuroinflammation in our LS mouse model. In this regard, daily CBD treatment (100 mg/kg) reduced both GFAP (Fig. 3a, b) and IBA1 staining (Fig. 3c, d) in the external globus pallidus (GPe), a key region susceptible to Ndufs4 deficiency23. Moreover, expression of TMEM119, a marker of homeostatic microglia38, was reduced in Gad2:Ndufs4cKO mice and its levels were normalized upon CBD treatment (Fig. 3e). Conversely, CBD did not affect GFAP, IBA1, or TMEM119 levels in control mice (Fig. 3a–e) in contrast to drugs tested in previous studies35,36. Subsequent 3D microglial morphological analyses revealed that microglia from Gad2:Ndufs4cKO mice suffered a morphometric remodeling to a rounded and larger perikarya with shorter processes, which was sensitive to CBD treatment (Fig. 3f–j). Notably, CBD treatment normalized the increased microglial soma volume observed in Gad2:Ndufs4cKO compared to control mice (Fig. 3f, g). This larger soma size observed in mutant mice was accompanied by a reduction in branch levels as well as fewer branches in each level (Fig. 3h), overall indicating an activated microglial state compared to the resting phenotype found in control mice. Remarkably, these alterations were also restored by CBD treatment (Fig. 3h). In contrast, no significant treatment differences were found in dendrite volume (Fig. 3i) and filament length (Fig. 3j). Taken together, these results indicate that daily CBD treatment normalizes morphometric microglial changes observed in an affected brain area of a LS mouse model.

Representative immunofluorescence images (left) and z-stack of higher magnification images (right) (a) and quantification of GFAP fluorescence signal intensity (b) of the astroglial marker GFAP in the GPe of daily CBD- or vehicle-treated control (Gad2:Ndufs4cCT) and Gad2:Ndufs4cKO mice (n = 9 [control-vehicle], 7 [control-CBD], 8 [Gad2:Ndufs4cKO-treated groups], two-way ANOVA/Tukey’s multiple comparisons test, ****p < 0.0001 [genotype effect]; ###p = 0.0008 [treatment effect]). Representative immunofluorescence images (left) and z-stack of higher magnification images (right) (c) and quantification of IBA1 fluorescence signal intensity (d) of the microglia/macrophage marker IBA1 in the GPe of daily CBD- or vehicle-treated Gad2:Ndufs4cCT and Gad2:Ndufs4cKO mice (n = 9 [control-vehicle], 7 [control-CBD], 8 [Gad2:Ndufs4cKO-treated groups], two-way ANOVA/Tukey’s multiple comparisons test, ***p = 0.0002 [genotype effect]; ###p = 0.0002 [treatment effect]). e Quantification of TMEM119 fluorescence signal intensity in the GPe of daily CBD- or vehicle-treated control and Gad2:Ndufs4cKO mice (n = 9 [control-vehicle], 7 [control-CBD], 8 [Gad2:Ndufs4cKO-treated groups], two-way ANOVA/Tukey’s multiple comparisons test, *p = 0.0446 [genotype effect]; ###p = 0.0006 [treatment effect]). f Representative 3D microglia reconstruction using Imaris of each group. g Quantification of soma volume/cell of IBA1+ cells (n = 9 [control-vehicle], 7 [control-CBD and Gad2:Ndufs4cKO-vehicle], 8 [Gad2:Ndufs4cKO-CBD], two-way ANOVA/Tukey’s multiple comparisons test, ****p < 0.0001 [genotype effect]; ####p < 0.0001 [treatment effect]). h Distribution plot of microglial ramification (n = 9 [control-vehicle], 7 [control-CBD and Gad2:Ndufs4cKO-vehicle], 8 [Gad2:Ndufs4cKO-CBD], two-way ANOVA/Tukey’s multiple comparisons test, ****p < 0.0001 [genotype effect]; #p = 0.0477, ###p = 0.0001 [treatment effect]). i Quantification of total dendrite volume/cell of IBA1+ cells (n = 9 [control-vehicle], 7 [control-CBD and Gad2:Ndufs4cKO-vehicle], 8 [Gad2:Ndufs4cKO-CBD], two-way ANOVA/Tukey’s multiple comparisons test, **p = 0.0013 [genotype effect]). j Quantification of total dendrite length/cell of IBA1+ cells (n = 9 [control-vehicle], 7 [control-CBD and Gad2:Ndufs4cKO-vehicle], 8 [Gad2:Ndufs4cKO-CBD]). All data are presented as mean values ± SEM. All source data are provided as a source data file. Veh vehicle, CT control mice, cKO Gad2:Ndufs4cKO mice. Scale bars: a, c: 100 μm (left) and 20 μm (right), f: 8 μm.
CBD does not normalize altered mitochondrial complexes assembly or respiration
NDUFS4 is also involved in complex I (CI) assembly and stability both in humans and mice10. Thus, we next analyzed respiratory holocomplexes in digitonin solubilized brain mitochondria of Ndufs4KO and control mice treated with vehicle or CBD (100 mg/kg/day) by first-dimension blue native gel electrophoresis (1D-BNGE). BNGE in-gel activities of CI and CI-containing supercomplexes uncovered almost inexistent signal in Ndufs4KO mice treated with vehicle or CBD (Supplementary Fig. S3a–c). Moreover, western blot analysis using antibodies against CI-subunit NDUFS3 in Ndufs4KO mice39 confirmed the presence of catalytically inactive 830 kDa assembly intermediates and the absence of CI holocomplex and CI supercomplexes. These results were unaffected by treatment with CBD (Supplementary Fig. S3c). Additionally, no differences between genotypes or treatments were observed in complex IV BNGE in-gel activities (Supplementary Fig. S3d).
We next sought to analyze whether the beneficial effects of a CBD treatment could also arise from a direct effect on mitochondrial function. To rule out a masking effect of mitochondria from healthy cells, we used mitochondrial preparations from the olfactory bulb of Ndufs4KO mice in which Ndufs4 is absent in all cells. Mitochondrial oxygen consumption rate (OCR) was assessed by using a Seahorse XFp extracellular flux analyzer. In the absence of any inhibitors, mitochondria from Ndufs4KO mice displayed a substantial decrease in the OCR (basal respiration) compared to control mice (Supplementary Fig. S4a, b) regardless of the treatment. A decreased OCR was also observed in Ndufs4KO mice after addition of ADP (state III), after ATP synthesis inhibition with oligomycin (state IV, proton leak), when the chemical uncoupler FCCP was subsequently injected to induce maximal respiration (state IIIu), and finally when antimycin A was added to assess non-mitochondrial respiration (Supplementary Fig. S4a, b). However, this decrease in OCR was not altered by CBD treatment, suggesting that CBD has no direct effect on respiration in mitochondria isolated from Ndufs4KO mice.
CBD’s in vivo mechanism does not appear to involve cannabinoid receptor modulation
We sought to assess the potential contribution of cannabinoid receptor(CBR)-related mechanisms in CBD’s beneficial effect15,40. First, administration of the CB1R agonist Δ9-tetrahydrocannabinol (THC)18 did not have any affect in Gad2:Ndufs4cKO lifespan (Supplementary Fig. S4c). Subsequently, to validate the lack of mtCB1R-mediated respiratory rescue due to CBD treatment, we performed OCR analysis with increasing doses of CBD or the full, selective CB1R agonist WIN-55212-241 in control and constitutive CB1R-KO42 mice. As expected, WIN-55212-2 robustly decreased CI-driven OCR (Supplementary Fig. S4d). CBD showed a clear biphasic response in both control and CB1R-KO (Supplementary Fig. S4d, e). In control animals, significant increase and decrease in OCR were only observed at the lowest (0.1 μM) and highest (100 μM) concentrations, respectively—while CBD had no significant effect at the concentration ranges tested for Ndufs4KO animals (1–10 μM). CB1R-KO mitochondria also showed significant OCR increase due to the lowest CBD concentration, with a comparable yet non-significant decrease at the highest concentration. We next assessed mtCB1R protein levels in immunoprecipitated mitochondria from Ndufs4KO animals (Supplementary Fig. S5a–d). Western blot indicated that the majority of CB1R in the olfactory bulb was not associated to mitochondria (Supplementary Fig. S5e). MtCB1R levels trended upwards in Ndufs4KO compared to control but did not reach significance (Supplementary Fig. S5f–h).
Furthermore, we determined the effect of Ndufs4 deficiency on CBR expression at the transcript level in the olfactory bulb of LS mouse models. The CB1R-encoding transcript, Cnr1, was moderately yet significantly increased in Ndufs4KO (Supplementary Fig. S6a). Contrarily, we observed a large increase in Cnr2 (Supplementary Fig. S6b) likely due to enhanced microglial reactivity. Assessment of the orphan GPCR, GPR55, thought to be a third putative CBR40, indicated no difference in its transcript (Gpr55) levels in Ndufsc4KO olfactory bulb (Supplementary Fig. S6c). Increased Cnr1 expression was not observed in Gad2:Ndufs4KO, while Cnr2 was again significantly upregulated (Supplementary Fig. S6d, e). No changes in Gpr55 expression were found (Supplementary Fig. S6f). Total CB1R protein was also not significantly altered in Gad2:Ndufs4cKO (Supplementary Fig. S7a).
Finally, we evaluated some of the key pathways cited as downstream effectors of CB1R activation43 in the olfactory bulb of Gad2:Ndufs4cKO mice. We did not observe any changes in phosphorylation levels of the extracellular signal-related kinase p44/p42 (ERK1/2) due to genotype or CBD treatment (Supplementary Fig. S7b, c). Changes in mTORC-related signal kinase cascades are involved in the cellular response to both CB1R activation and whole-body Ndufs4KO22,44,45. Thus, we probed the activation status of its related signaling kinases. No significant changes were observed in the phosphorylation levels of the serine/threonine kinase AKT1. The ribosomal protein S6 phosphorylation (Supplementary Fig. S7d, e) was decreased by genotype and treatment (p < 0.05), but neither the factor interaction nor individual intergroup comparisons reached significance. Overall, these results indicate that CBR expression and signaling may be altered by NDUFS4 deficiency. However, they do not appear to be involved in the beneficial effect of CBD in the models tested.
PPARγ blockade reduces CBD-induced beneficial effects in complex I deficient mice
We next set to elucidate potential pharmacological targets of CBD involved in the beneficial effects reported here37. In this regard, PPARγ has been widely reported to be a key player in mitochondrial function and neurological diseases46, as well as an important component mediating CBD’s beneficial effects37,47. To determine whether PPARγ was involved in the CBD-mediated effects, Gad2:Ndufs4cKO mice were pretreated with the PPARγ antagonist GW9662 (1 mg/kg) 20 min before each CBD (100 mg/kg) or vehicle administration twice daily, beginning before the period of seizure risk (PND33-37). Notably, CBD-induced lifespan extension in Gad2:Ndufs4cKO mice was significantly reduced with GW9662 pretreatment (Fig. 4a).

a Kaplan–Meier survival curve of Gad2:Ndufs4cKO mice treated daily with vehicle (n = 24, gray), CBD (n = 18, red), GW9662+Veh (n = 16, blue), GW9662 + CBD (n = 18, green), and control mice (n = 20, black). GW9662 (1 mg/kg) was administered 20 min before each CBD or vehicle injection (Long-rank test, ****p < 0.0001 vs Veh-treated Gad2:Ndufs4cKO mice; ##p = 0.0040 vs CBD-treated Gad2:Ndufs4cKO mice; &p = 0.0112 vs GW9662-Veh-treated Gad2:Ndufs4cKO mice). Survival graphs of Gad2:Ndufs4cKO-Veh (gray) and Gad2:Ndufs4cKO-CBD (red) are also displayed in Fig. 2a. % of time spent in open arms (b), % of entries in open arms (c), and total arm entries (d) in the elevated plus maze of Gad2:Ndufs4cKO and control mice pretreated daily with GW9662 20 min before vehicle (blue, n = 16 [control], 15 [Gad2:Ndufs4cKO]) or CBD (green, n = 11 [control], 18 [Gad2:Ndufs4cKO]) administration (two-way ANOVA followed by Tukey’s multiple comparisons test, ****p < 0.0001 vs Veh-treated control; &&p = 0.0028, &&&p = 0.0002 vs CBD-treated control). Discrimination index of the time exploring a stranger mouse over an empty wire cage (e) and total exploration time (f) in the 3-chamber social interaction test of Gad2:Ndufs4cKO and control mice pretreated daily with GW9662 20 min before vehicle (blue, n = 14 [control], 18 [Gad2:Ndufs4cKO]) or CBD (green, n = 9 [control], 24 [Gad2:Ndufs4cKO]) administration (two-way ANOVA followed by Tukey’s multiple comparisons test, *p = 0.0178, **p = 0.0010 vs Veh-treated control; &p = 0.0322, &&p = 0.0048 vs CBD-treated control). Quantification of IBA1 (g) or GFAP (h) fluorescence signal intensity in the GPe of Gad2:Ndufs4cKO and control mice pretreated daily with GW9662 20 min before vehicle (blue, n = 6 [control], 4 [Gad2:Ndufs4cKO]) or CBD (green, n = 6) administration (one-way ANOVA followed by Tukey’s multiple comparisons test, *p = 0.0404, ***p = 0.0007 vs Veh-treated control; #p < 0.05 vs Veh-treated cKO). All data are presented as mean values ± SEM. All source data are provided as a source data file. Veh vehicle, CT control mice, cKO Gad2:Ndufs4cKO mice.
We next assessed whether PPARγ also contributed to the CBD-induced improvement in behavioral comorbidities. In the elevated plus maze, the pretreatment with GW9662 blunted CBD effect on Gad2:Ndufs4cKO mice regarding the percentage of time spent in open arms (Fig. 4b) and the percentage of entries in open arms (Fig. 4c). No differences in total arm entries were found between either genotypes or treatments (Fig. 4d). A similar effect was observed in the 3-chamber social interaction test, where GW9662 pretreatment prevented the rescue of social deficits induced by CBD (Fig. 4e, f), suggesting that CBD largely mediates its effects via PPARγ. In contrast, the reduction in intensity signal of the microglial marker IBA1 (Fig. 4g) and the astrocytic marker GFAP (Fig. 4h) induced by CBD was not prevented by GW9662 pretreatment, indicating that CBD reduces neuroinflammation in the GPe of this LS mouse model via a PPARγ−independent mechanism.
The PPARγ agonist leriglitazone mimics CBD’s beneficial effects
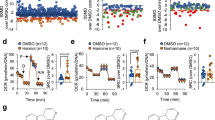
To validate the crucial role of PPARγ in the CBD-induced amelioration of LS pathology, Ndufs4KO and Gad2:Ndufs4cKO mice received oral leriglitazone, a novel brain penetrant PPARγ agonist with an improved profile for CNS diseases48,49. Like CBD, daily exposure to leriglitazone significantly extended lifespan in Ndufs4KO (Fig. 5a) and in Gad2:Ndufs4cKO mice (Fig. 5b). Additionally, leriglitazone delayed the appearance of clinical signs such as curling and clasping (Fig. 5c) as well as the motor decline observed in Ndufs4KO mice in the rotarod test (Fig. 5d). Daily leriglitazone treatment also moderately decreased tidal volume (Fig. 5e) and increased respiratory frequency compared with Ndufs4KO mice (Fig. 5f). However, in contrast to CBD, no differences were found in total ventilation (Fig. 5g). Like CBD, leriglitazone did not have any effect on respiratory rates from mitochondria isolated from the olfactory bulb of Ndufs4KO mice (Fig. 5h, i). Overall, the PPARγ agonist leriglitazone prolongs survival and ameliorates disease comorbidities similarly to CBD without having a direct effect on mitochondrial respiration. Given the mechanism of action of leriglitazone, in addition to a neuroprotective action, anti-inflammatory effects in specific brain regions through repression of the nuclear factor B (NF-kB) pathway could be driving the beneficial effects observed in the mice.

a Kaplan–Meier survival curve of Ndufs4KO mice exposed to regular (n = 8, gray) or leriglitazone-containing food (n = 9, blue, Log-rank test **p = 0.0048), and control mice (n = 20, black). b Kaplan–Meier survival curve of Gad2:Ndufs4cKO mice exposed to regular food (n = 10, gray) or leriglitazone-containing food (n = 12, blue, Log-rank test **p = 0.0045), and control mice (n = 20, black). c Clasping/twisting onset in Ndufs4KO mice exposed to regular (n = 11, gray) or leriglitazone-containing food (n = 12, blue, unpaired two-tailed t-test, **p = 0.0026). d Rotarod performance in Ndufs4KO exposed to regular (n = 11, gray) or leriglitazone-containing food (n = 12, blue, two-way ANOVA followed by Sidák’s multiple comparisons test, *p = 0.0313 treatment effect). Time to fall off is represented among the 3 tested days (PND 30, 40, and 50). Respiratory tidal volume (e), frequency (f), and total ventilation (g) of Ndufs4KO and control mice exposed to regular or leriglitazone-containing food (n = 14 [control groups], 10 [Ndufs4KO-regular], 11 [Ndufs4KO-leriglitazone], two-way ANOVA followed by Tukey’s multiple comparisons test, **p = 0.0018, ****p < 0.0001 [genotype effect]; #p < 0.05 [treatment effect]). Boxplots show the median (horizontal line), upper and bottom quartiles (box outlines) and the maximum and minimum non-outlier data points (whiskers). h Representative oxygen consumption rate (OCR) curves of purified mitochondria from the olfactory bulb of control (empty circles) and late-stage disease Ndufs4KO mice (filled circles) that were daily exposed to regular (gray) or leriglitazone-containing food (blue). OCR was normalized to the total protein amount. i OCR quantification from Fig. 5h, including: basal respiration, after adding ADP (state III), after adding the ATP synthase inhibitor oligomycin (proton leak), after adding the uncoupler FCCP (maximal), and after adding the cytochrome C inhibitor antimycin A (non-mito) (n = 7 [control-regular], 4 [Ndufs4KO-regular], 6 [leriglitazone groups], two-way ANOVA followed by Tukey’s multiple comparisons test, *p = 0.0299, **p = 0.0084, ****p < 0.0001 [genotype effect]). All data are presented as mean values ± SEM. All source data are provided as a source data file. Veh vehicle, CT control mice, KO Ndufs4KO mice, cKO Gad2:Ndufs4cKO mice, VT Tidal volume, fR Respiratory frequency, VE Total ventilation, Reg Regular, Leri Leriglitazone.
PPARγ expression is reduced in LS mouse models and in patient-derived fibroblasts
The beneficial effects of PPARγ activation by either CBD or leriglitazone pointed to a key role of this receptor in the pathophysiology of LS, thus uncovering a novel target to study. We next sought to assess whether PPARγ expression was conserved both in LS mouse models and in patient-derived fibroblasts. Hence, we first performed qRT-PCR of Pparg mRNA levels in two affected areas of late-stage (~PND45-50) Ndufs4KO mice, namely the olfactory bulb and cerebellum22. Notably, we found a significant decrease in Pparg expression in mutant mice compared to control only in the olfactory bulb (Fig. 6a) but not in the cerebellum (Supplementary Fig. S8a), suggesting a brain region-specific altered expression of this receptor. Similarly, reduced Pparg mRNA levels were also present in the olfactory bulb of Gad2:Ndufs4cKO mice compared to control mice (Fig. 6b), while expression levels were unaltered in the unaffected areas of the cerebellum, brainstem, striatum (Supplementary Fig. S8b–d). To parse whether Pparg levels were reduced in the cell population bearing Ndufs4 deletion –namely, Gad2-positive neurons– or if it was a consequence of the triggered inflammatory response (i.e. microgliosis and astrogliosis) we performed RiboTag-Seq50 to isolate transcripts exclusively in GABAergic cells (Fig. 6c). Our approach revealed a significant decrease in Pparg levels in GABAergic neurons from the olfactory bulb of mutant compared to control mice (Fig. 6d). In contrast, GABAergic cells similarly isolated from the unaffected cerebellum in Gad2:Ndufs4cKO showed no changes in Pparg levels (Supplementary Fig. S8e).

a qRT-PCR for Pparg mRNA expression normalized with Gapdh in the olfactory bulb from control and Ndufs4KO mice (n = 10 mice/group, unpaired two-tailed t-test, ****p < 0.0001). b qRT-PCR for Pparg mRNA expression normalized with Gapdh in the olfactory bulb from control (n = 8 mice/group) and Gad2:Ndufs4cKO mice (n = 8 mice/group, unpaired two-tailed t-test, *p = 0.0111). c Schematic representation of the RiboTag approach to isolate mRNAs from Gad2-positive cells. d qRT-PCR for Pparg mRNA expression normalized with Gapdh after HA-tagged-ribosome immunoprecipitation from Gad2-positive cells in the olfactory bulb from control (n = 6) and Gad2:Ndufs4cKO:RiboTag mice (n = 7, unpaired two-tailed t-test, **p = 0.0060). Luciferase activity assay in primary neuronal (e) and astrocytic (f) cultures from control and Ndufs4KO mice that were transfected with the reporter plasmid PPRE-Luc (n = 4 replicates/condition; unpaired two-tailed t-test, ***p = 0.0002). g qRT-PCR for PPARG mRNA expression normalized with GAPDH in control (n = 8 replicates/condition), NDUFS4-mutant (n = 3 replicates/condition) and COX15-mutant (n = 4 replicates/condition) from 3-independent cultures of patient-derived skin fibroblasts (relative expression with respect to the mean value of controls; one-way ANOVA followed by Dunnett’s multiple comparisons test, *p = 0.0343). h Western Blot analysis of PPARγ1/2, NDUFS4, COX5A, and GAPDH expression levels in control, NDUFS4- and COX15-mutant patient-derived skin fibroblasts (lower panel) and quantification of PPARγ1 normalized to GAPDH (upper panel) (n = 3 replicates/group, one-way ANOVA followed by Dunnett’s multiple comparisons test, ***p = 0.0010). All data are presented as mean values ± SEM. All source data are provided as a source data file. CT control mice, KO Ndufs4KO mice, cKO Gad2:Ndufs4cKO mice.
To assess whether PPARγ activity was also altered, we performed primary neuronal and astrocytic cultures from Ndufs4KO mice and wildtype littermates. PPARγ acts as a transcription factor and to measure its activity we transfected cultured neurons with the PPARγ response element-coupled to luciferase (PPER-Luc). The luciferase assay revealed a 55% decrease in PPARγ activity in cultured neurons from Ndufs4KO mice compared to those from wildtype littermates (Fig. 6e), but not in primary astrocyte cultures (Fig. 6f). In contrast to the results obtained in vivo, similar Pparg transcript levels were found in both genotypes of the neuronal cultures (Supplementary Fig. S8f). These results suggest that Pparg expression or activity are reduced only in the affected neuronal populations of two LS mouse models.
We next assessed PPARγ expression levels in patient-derived fibroblasts. To do so, we used skin fibroblasts from a LS patient bearing NDUFS4 mutation51 –akin to the two Ndufs4-mutant mouse lines used in the present study. In addition, we included fibroblasts from another LS patient harboring a mutation in COX15, a gene encoding for a protein involved in heme A biosynthesis, necessary for complex IV assembly. We measured PPARG levels in fibroblasts from these two LS patients and two commercially available pediatric fibroblasts from healthy individuals by qRT-PCR. Our analysis revealed a 50% decrease in PPARG expression in both NDUFS4 and COX15-mutant cells compared to control fibroblasts (Fig. 6g). Furthermore, we report a significant decrease in PPARγ protein expression in both NDUFS4 and COX15-mutant fibroblasts (Fig. 6h), suggesting that PPARγ downregulation may be a conserved mechanism contributing to the pathophysiology of LS.
CBD improves the phenotype of LS patient-derived fibroblasts
Given the significant beneficial effects of CBD in the two LS phenotypic presentations used in this study, we next assessed whether CBD could also exert a favorable effect in patient-derived fibroblasts. NDUFS4-mutant skin fibroblasts show a decreased proliferation compared to control fibroblasts (Fig. 7a) as previously reported51. We then treated NDUFS4-mutant fibroblasts with CBD (5 µM) or its vehicle and we measured proliferation (% RFU) 24 h later. As we noticed that the proliferation rates decreased proportionally to the number of passes the fibroblasts went through (see the disperse variability in Fig. 7a), we plotted the paired comparisons between vehicle- and CBD-treated fibroblasts of each independent biological replicate and we observed an overall increased proliferation induced by CBD (Fig. 7b, p = 0.011). However, repeated exposure to 5 µM CBD for 3.5 days induced a robust decrease in proliferation (Supplementary Fig. S9a), highlighting the importance of dosage regimen in cell culture studies. On the other hand, a lower dose (0.1 µM) still increased proliferation rates in mutant compared to control fibroblasts after a repeated 3.5-day exposure while a dose of 0.5 µM had no effect (Supplementary Fig. S9a).

a Cell proliferation measured as values of relative fluorescence units (RFU) for control and NDUFS4-mutant patient-derived fibroblasts. Values are represented as % of RFU respect to one of the two control fibroblast lines used in this study. Each value is the mean of each biological triplicate/experiment (n = 5 [control], 6 [NDUFS4], one-way ANOVA/Tukey’s multiple comparisons test, **p = 0.0012). b Cell proliferation measured as values of relative fluorescence units (RFU) for NDUFS4-mutant patient-derived fibroblasts treated with vehicle or CBD (5 µM) for 24 h. Each value is the mean of each biological triplicate/experiment (n = 6 independent experiments/condition, paired two-tailed t-test, *p = 0.0111). The average value of each group is represented in brown. Heatmap (c) and volcano plot (d) of the differentially expressed genes between LS patient-derived fibroblasts (both NDUFS4 and COX15 mutations) and two commercial control fibroblasts treated with vehicle for 24 h. Expression values are color coded according to the legend. The dendrogram depicts hierarchical clustering. e Volcano plot of differentially expressed genes between vehicle- and CBD-treated (5 µM) fibroblasts for 24 h. f Immunofluorescence of Mitotracker (cyan) and MitoSOX (magenta) probes, and nuclear cell staining (DAPI, blue) in NDUFS4-mutant fibroblasts. Note the colocalization of both probes. Scale bar: 20 μm. g Quantification of MitoSOX staining in COX15- and NDUFS4-mutant fibroblasts as well as in two control fibroblasts (CT1 and CT2) treated with vehicle or CBD (1 µM) for 24 h (n > 560 cells over three biological independent experiments, two-way ANOVA/Tukey’s multiple comparisons test, ****p < 0.0001 [genotype effect]; ##p < 0.01, ####p < 0.0001 [treatment effect]). h Quantification of total glutathione levels in COX15- and NDUFS4-mutant fibroblasts as well as in two control fibroblasts (CT1 and CT2) treated with vehicle or CBD (1 µM) for 24 h (n = 4 technical replicates for all groups, except CT1-vehicle with n = 5, over three biological independent experiments; two-way ANOVA/Tukey’s multiple comparisons test, #p = 0.0109, ####p < 0.0001 [treatment effect]). All data are presented as mean values ± SEM. All source data are provided as a source data file. GSH Glutathione, Veh vehicle.
To gain more insights about CBD’s mechanisms of action that might explain its beneficial effects, we performed RNAseq of LS patients-derived fibroblasts and control cells treated with CBD (5 µM) or vehicle for 24 h. We included both NDUFS4- and COX15-mutant fibroblasts to identify differentially expressed genes common to both LS-related mutations. As expected, both mutant skin fibroblast lines clustered together in comparison with two control fibroblast lines (Fig. 7c), highlighting common gene dysregulation in two LS patients despite bearing mutations in genes involved in different respiratory complexes (CI for NDUFS4 and CIV for COX15). Our RNAseq data revealed 5361 dysregulated protein-coding genes between vehicle-treated mutants and control fibroblasts. Among them, 2658 genes were upregulated, and 2703 genes were downregulated (Fig. 7d and Supplementary Data 1). However, when comparing vehicle- versus CBD-treated fibroblasts, only 5 protein-coding genes were significantly upregulated (Fig. 7e and Supplementary Data 2). The 3 genes with the highest fold change encode for metallothioneins (MT1X, MT1E, and MT2A), which have been widely reported to function as antioxidants against oxygen and nitrogen species52. The fourth largest increased gene expression induced by CBD was SLC3A2, which has also been related with oxidative processes53. As exacerbated ROS levels typically accompany mitochondrial dysfunction, these results raise the possibility that alteration of oxidative processes might be key to CBD benefit in the LS models tested here.
CBD attenuates ROS and upregulates antioxidant defenses in LS patient-derived fibroblasts
Given the distinct antioxidant defense mechanisms implied by the RNASeq results described above, we analyzed mitochondrial ROS levels in LS patient-derived fibroblasts. We first verified our approach by showing a colocalization of MitoSOX and Mitotracker probes, which detect mitochondrial ROS levels and mitochondria respectively, in NDUFS4-mutant fibroblasts (Fig. 7f). MitoSOX response was validated by treating control cells with rotenone, a selective CI inhibitor that reliably induces CI-driven ROS production54 (Supplementary Fig. S10a, b). We next incubated mutant and control fibroblasts with CBD (1 µM) or vehicle for 24 h and we measured the intensity of MitoSOX staining. First, within vehicle-treated groups, we found elevated ROS levels in both COX15- and NDUFS4-mutant fibroblasts, being higher in the latter, compared to two control fibroblasts (Fig. 7g, Supplementary Fig. S10c). CBD exposure significantly reduced ROS levels in all experimental groups (Fig. 7g, Supplementary Fig. S10c). These results suggest that CBD exerts therapeutically relevant antioxidant effects in LS patient-derived cells.
To validate the upregulation of MT1/2 transcripts in vivo, their expression was quantified in primary neuronal cultures from Ndufs4KO mice. As in the fibroblasts, CBD significantly upregulated Mt1 and Mt2 levels in neurons (Supplementary Fig. S10d, e). However, the Slc3a2 and Chst2 increases were not replicated in this context (Supplementary Fig. S10f, g). SLC3A2 is associated with the cysteine transporter that fuels glutathione synthesis and CBD has been shown to upregulate this process55, thus we quantified total glutathione levels in patient fibroblasts. CBD slightly upregulated glutathione levels in controls, while restoring levels in both NDUFS4 and COX15 mutants towards control levels (Fig. 7h). Altogether, these results suggest that CBD exerts potent antioxidant activity by upregulating MT and glutathione-related ROS defense systems –although not all elements are equally conserved between mice and humans.
Discussion
Here we demonstrate that daily CBD treatment prolongs lifespan and attenuates disease progression in two in vivo phenotypic presentations of LS and improves patient-derived cells’ phenotype. Although various preclinical interventions could better ameliorate disease onset/severity and brain pathology in Ndufs4KO mice compared to what we report for CBD, none have been approved to date. Treatments that showed preclinical promise such as reduction of oxygen bioavailability56,57, rapamycin22, doxycycline58, pexidartinib36, and ruboxistaurin35 (see ref. 10 for an extended review) are hindered by various drug adverse and off-target effects, and/or practical concerns, which limit their translational potential. For instance, while rapamycin potently increases survival of Ndufs4KO upon pre-symptomatic treatment initiation, we found that rapamycin does not extend lifespan when initiated at the same, mid-stage age as CBD (PND35-40). This emphasizes the translational potential of CBD, with a potentially much larger time window for effective treatment onset. Of note, the use of CBD for clinical applications has gained much attention given its high-benefit-to-risk ratio and lack of psychoactive properties15,17,18,19,20,21, the latter being especially important in the context of pediatric diseases such as LS. Therefore, the safe and well-tolerated profile of CBD21 in combination with the substantial beneficial effects found in this study point to this cannabinoid as a potential effective treatment for LS patients.
CBD effectively prevents fatal seizures in an MD-mediated fatal epilepsy mouse model (Gad2:Ndufs4cKO mice)23, in agreement with its known anticonvulsant properties in models of acute seizure, chronic epilepsy and epileptogenesis59. Epilepsy is a common clinical manifestation of MDs including LS, and its incidence in mitochondrial encephalopathy varies between 35 and 61%60. Common antiepileptic drugs such as sodium valproate are not recommended in MD due to their mitochondrion-toxic effects61, while marked resistance to other first-line treatments such as carbamazepine, levetiracetam, and perampanel has also been characterized in patients and Gad2:Ndufs4cKO mice23. Notably, the anticonvulsant doses found in this study are in agreement with the effective dosages described in other mouse models of treatment-resistant epilepsies, such as Angelman syndrome62 and Dravet syndrome28, highlighting the potential translatability of our current results. Together, our findings suggest that CBD might reduce seizures in LS patients with a tolerable toxicity as reported for other pediatric indications21, thereby extending the therapeutic spectrum of CBD’s anticonvulsant effects.
Our results expand the list of neurodegenerative conditions wherein CBD exerts antioxidant and anti-inflammatory actions15 to include LS. Daily CBD administration was sufficient to dampen neuroinflammation in the GPe, a nucleus of the basal ganglia found to be highly affected in our MD-fatal epilepsy mouse model23 as well as in LS patients4,5,6. This agrees with the anti-inflammatory and immune suppressive role of CBD in several neurodegenerative and autoimmune diseases20. Similarly, previous MD interventions that reduced inflammation, such as rapamycin22 or doxycycline58, and either leukocyte36 or microglial depletion63 could also extend survival and improve the phenotype in Ndufs4KO. Furthermore, we found that CBD reduces ROS levels in LS patient-derived fibroblasts. This could be due to its potent, inherent antioxidant effect or its ability to bolster antioxidant defenses37,64. We provide evidence for the latter argument, showing that CBD increases metallothionein expression in both LS patient fibroblasts and Ndufs4KO neurons, as has been described for LPS-challenged microglia55. MTs are involved in the detoxification of heavy metals and various free radical species, and their expression levels have previously been related to epilepsy in mice52. High levels of MT1/MT2 are correlated to decreased neuronal death following seizures, while Mt2-deficient mice display increased ROS, neuronal death and seizures following kainic acid treatment52. Future in vivo studies are required to determine whether CBD might exert antiseizure effects by upregulating MT1/2 in mitochondrial epilepsies. Contrarily, metallothionein overexpression alone does not improve the Ndufs4KO phenotype65, emphasizing the inability of a single antioxidant mechanism to maintain ROS homeostasis in LS.
In that regard, our results show that CBD also upregulates SLC3A2, a protein that is associated with the cystine transporter SLC7A11. This complex is critical for the synthesis of the antioxidant glutathione (GSH)53, as evidenced by reduced GSH and increased oxidative stress in Slc3a2-deficient mice53. Accordingly, CBD treatment upregulates total GSH levels in all LS fibroblast lines tested. Despite the analytical limitations in quantifying GSH dynamics66, metabolomics evidence implicates dysregulated GSH homeostasis in various MD models and patients67. In Ndufs4 mutations specifically, an increased GSH pool and diminished GPx activity have been reported68,69, although discrepancies between studies necessitate further validation. The different baseline GSH levels found with the different LS genetic variants in this study illustrate that care must be taken in extrapolating these results to other MD. Critically, single antioxidant treatments have shown limited effectivity against LS in various clinical and preclinical studies10,70, underscoring the potential of treatments such as CBD that provide benefit through engaging multiple therapeutic targets. Together, our results show that CBD exerts therapeutic action through various antioxidant and anti-inflammatory pathways in parallel.
Our results indicate that the effective CBD dose used here has no direct effect on mitochondrial respiration or complex stability, consistent with previous interventions showing benefit without restoring CI assembly and activity22,57,58. CBD is thought to exert its therapeutic actions through multiple protein targets15,37. Although CBD has validated (low potency) inverse agonist or negative allosteric modulator activity at CB1R, our results support previous evidence that CBD benefit is far more likely mediated by non-cannabinoid mechanisms15,37. THC administration within the same setup as CBD did not extend lifespan in the models tested and extensive investigation of CB1R/mtCB1R expression in response to Ndufs4KO, Gad2:Ndufs4cKO and CBD treatment did not show any significant alterations. The upregulation of CB2R expression in affected brain tissues of Ndufs4KO mice is likely due to increased microglial expression thereof, as is frequently reported in neurodegenerative conditions71. While links between glial CB2R, CBD treatment and attenuated neuroinflammation exist, future studies will be needed to define its role in context of MD. Taken together, this study suggests that the CBD benefit in LS is most likely mediated by inflammatory and oxidative processes that do not involve rescue of CI structure/function or cannabinoid receptor interactions.
We also uncover a downregulation of PPARγ in a highly affected brain area of both Ndufs4-related mouse phenotypes as well as in patient-derived skin fibroblasts, suggesting that this may be a common feature in LS. PPARγ is a ligand-activated transcription factor that regulates expression of genes involved in redox balance, immune responses, metabolism, mitochondrial function, and other processes with high relevance in CNS-related diseases46. All these processes are highly compromised in LS, making PPARγ a promising target. The observed downregulation in Ndufs4KO compared to control mice suggests that decreased Pparg levels might be a feature of GABAergic cells in the highly affected regions where these neurons predominate. Despite the unchanged Pparg levels observed in three unaffected brain regions (including cerebellum), we found a reduction in PPARγ activity in cerebellar neurons. This suggests that PPARγ effects are compromised in Ndufs4-deficient mice, albeit with cell-type specific differences. From a translational standpoint, we also found a significant PPARG downregulation in fibroblasts derived from individuals with LS caused by mutations in two different genes (NDUFS4, COX15). Future studies are required to evaluate whether decreased PPARG expression is conserved in other MDs and whether an increase in PPARγ activity could improve the phenotype in these conditions.
In the present study, we uncovered PPARγ as one of the main receptors mediating the beneficial effects of CBD administration in Gad2:Ndufs4cKO mice. While the therapeutic promise of full PPARγ agonists is being revisited in light of refutations of their previously described toxicity72,73,74, therapeutic interest still majorly remains with partial ligands. CBD can act as a low affinity PPARγ agonist15,37,75, as demonstrated by the beneficial effects of CBD-induced PPARγ activation20. Accordingly, we show that CBD-induced lifespan extension in Gad2:Ndufs4cKO mice is attenuated after pretreatment with a PPARγ antagonist. It is plausible that CBD prolongs survival of these mice by exerting anticonvulsant activities largely via PPARγ activation. Indeed, compelling evidence indicates the potential of targeting PPARγ in epilepsy, especially in high-risk and treatment-resistant cohorts (see ref. 76 for review). PPARγ expression levels also seem to play a key role, as ketogenic diet interventions have been shown to reduce seizures in pediatric refractory epilepsy, in part via the upregulation of the PPARγ2 isoform77. Similarly, the anticonvulsant effects of CBD in a temporal lobe epilepsy model have been associated with PPARγ upregulation in the hippocampus47.
In addition to survival extension, we show that PPARγ antagonism blunts the CBD-induced normalization of both social and anxiety-related behavior, highlighting the importance of this receptor in mediating CBD’s therapeutic effects. Overall, our data suggests that activation of PPARγ by CBD might reduce seizures and thereby prevent premature mortality and their associated comorbidities in Gad2:Ndufs4cKO mice. However, our results show that CBD reduces neuroinflammation in a PPARγ-independent manner, contrary to existing literature on CBD’s PPARγ-mediated anti-inflammatory effects in other CNS disorders78,79. As inflammation in pathogenic areas can be attenuated even though survival is not improved, this suggests that a decrease in the inflammatory response alone is not sufficient to robustly ameliorate the phenotype in these mice. Future studies should aim to substantiate this hypothesis by evaluating the correlation between PPARγ activity, survival and neuroinflammation, towards a comparison of the different anti-inflammatory and pro-survival mechanisms involved.
To further demonstrate the beneficial effect of PPARγ activation in LS, we tested the novel, selective PPARγ agonist leriglitazone in both LS model phenotypes. We found that daily exposure to leriglitazone prolongs lifespan in both mouse lines and improves the phenotype in Ndufs4KO—albeit to lesser extents than CBD. This is in agreement with a previous study showing that bezafibrate, a pan-PPAR agonist (although with low affinity for PPARγ), enhances survival and attenuates disease progression in Ndufs4KO mice80. In this regard, our results underscore the key role for PPARγ agonism in the amelioration of the pathology of Ndufs4KO mice. Leriglitazone specifically holds promise in CNS disorders, given its favorable brain penetration at lower doses than other glitazones49,81. However, CBD still outperformed leriglitazone in LS model mice, indicating that CBD may provide benefit through a combination of different pharmacodynamic mechanisms, including PPARγ. This raises the potential of synergy between CBD and low doses of PPARγ agonists in MD therapy, limiting toxicity and maximizing the benefit-to-risk ratio.
Overall, our findings identify an altered PPARγ expression/activity in LS patients and genetic mouse models and show that CBD-mediated activation of this pathway significantly ameliorates disease outcomes. However, key limitations of this study should be considered before translation to clinical trials. CBD benefit should be confirmed in models with other LS-causing mutations. The potential effect of CBD on mitochondrial parameters other than RC function and assembly (i.e. mitochondrial dynamics & stress responses) should be considered in future developments. A better understanding of the PPARγ dependent and independent mechanisms of CBD is needed, considering the implication of CB2R, GSH and MT1/2 in MD arising from, but not fully explored in this work. This would be particularly relevant to determining possible synergistic and adverse drug interactions in LS patients, that currently rely on multidrug palliative treatments.
Nevertheless, CBD is already FDA/EMA-approved for other rare pediatric indications which may streamline its development for MD. This holds significant therapeutic implications, as clinical translation in rare diseases is usually challenging. Taken together, our results point to CBD as a potential effective treatment for LS.
Methods
Animals
All animal experiments were performed with the approval of the ethical committee at Universitat Autònoma de Barcelona (CEEAH) and Generalitat de Catalunya (DMAH). Animals were housed under standardized conditions with a 12-h light/dark cycle, stable temperature (22 ± 2 °C), controlled humidity (55 ± 10%), and food (Global diet 2018 S, Envigo) and water ad libitum. The following mouse lines were used in this study: Gad2Cre/+ (GAD2-IRES-Cre) purchased from The Jackson Laboratory, Ndufs4lox/lox and Ndufs4Δ/+ generated by our group8,82, and RiboTag50 from The Jackson Laboratory. All mice were on a C57BL/6J background after backcrossing for at least 10 generations. Mice lacking Ndufs4 constitutively (Ndufs4KO) and control mice (including both wild-type and heterozygous littermates, referred collectively as control) were obtained as described8. Ndufs4KO mice were humanely euthanized according to the ethical committee protocol when body weight declined by more than 20% of maximum.
Conditional knockout mice lacking Ndufs4 selectively in GABAergic cells (Gad2:Ndufs4cKO) and control mice (Gad2:Ndufs4CT) were obtained as described23. Mice with Gad2Cre/+:Ndufs4lox/+ genotype with one functional Ndufs4 allele as well as mice with the Gad2+/+:Ndufs4lox/lox genotype were used as controls. These conditional knockout mice were also crossed with the Cre-dependent RiboTag mouse line to generate a triple transgenic mouse line that expresses HA-tagged-ribosomes selectively in Gad2-positive cells (Gad2Cre/+:Ndufs4lox/+:RiboTagHA/+ as Gad2:Ndufs4CT:RiboTag and Gad2Cre/+:Ndufs4lox/lox:RiboTagHA/+ as Gad2:Ndufs4cKO:RiboTag).
CB1-KO and their wilt-type littermates were bred and maintained as described42. The experimental procedures for these mice were approved by the Committee on Animal Health and Care of INSERM, and the French Ministry of Agriculture and Forestry (authorization number 3306369) and the French Ministry of Higher Education, Research and Innovation (authorization APAFIS#20053). Both male and female mice were included in all experiments, each separate measurement representing a single animal. Animal welfare and endpoint monitoring as well as euthanasia were performed by accredited vivarium staff and analysts, according to approved protocols of the UAB/INSERM. All reporting of animal experiments abides by the essential ARRIVE guidelines for animal research, as defined by the NC3RS (London, GB).
Drugs and treatments
(-)-Cannabidiol (CBD) and Δ9-tetrahydrocannabinol (THC) were purchased from THC Pharm-GmbH. Cremophor-EL, DMSO, PEG-400, Tween-80, WIN55.212-2 Mesylate salt and GW9662 were purchased from Sigma-Aldrich. Rapamycin was purchased from MedChemExpress. Leriglitazone (5-[[4-[2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]−1,3-thiazolidine-2,4-dione hydrochloride), which is in clinical development for adrenoleukodystrophy83, was provided by Minoryx Therapeutics. CBD and THC were diluted in vehicle preparation containing 5% ethanol (v/v), 5% Cremophor-EL (v/v), and 90% saline (v/v). Rapamycin was similarly diluted in a vehicle of 5% ethanol (v/v), 5% PEG-400 (v/v), 5% (v/v) Tween-80 and 85% saline (v/v). GW9662 was diluted in 5% DMSO (v/v) and 95% saline (v/v). CBD and vehicle solutions were administered 1 h prior to the behavioral task. GW9662 was administered 20 min before CBD/vehicle administration. Leriglitazone was administered in the food (AIN76A, Ssniff Spezialdiäten) at a concentration of 0.03% (equivalent to 50 mg/kg/day) as reported49, starting at ~PND25 until endpoint. The same diet product without leriglitazone was used as a control. All other drugs were administered intraperitoneally (i.p.) in a volume of 10 mL/kg.
Cultured skin fibroblasts and treatments
Patient skin fibroblasts were provided by Dr. Julio Montoya (Universidad de Zaragoza, Spain) and Dr. Miguel Ángel Martín (Hospital 12 de Octubre, Madrid, Spain) and used under approval 5393 from the Universitat Autònoma de Barcelona Ethics Committee. Informed consent was obtained from donors. Control human pediatric dermal fibroblasts were commercially obtained from Innoprot (P10856) and Cell Applications, Inc (106-05n). Primary mutant and pediatric control skin fibroblasts were grown in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (4.5 g/L glucose, 1 mM sodium pyruvate) supplemented with 10% (v/v) fetal bovine serum (GIBCO Thermo Fisher Scientific, #10270) and 1% (v/v) Pen/Strep (GIBCO Thermo Fisher Scientific, #15140) in an incubator at 37 °C with a humidified atmosphere at 5% CO2. Cells were treated with 0.1–5 µM CBD for 24/72 h. CBD stock solution was prepared in DMSO (Sigma-Aldrich, #D8418) with a final concentration of 0.1% (v/v) DMSO in cell culture. After treatment, cells were collected and used for assays. Cell lines were regularly tested for mycoplasma.
RNA extraction, RNAseq library preparation, and sequencing
For RNAseq, fibroblasts were treated with 5 µM CBD or vehicle for 24 h and total cellular RNA was extracted using the RNeasy Mini Kit (Qiagen, #74106) according to manufacturer’s instructions, followed by DNAse (RNase-Free DNase Set, Qiagen, #79254) treatment. The RNAseq library was prepared with KAPA Stranded mRNA-Seq Illumina Platforms Kit (Roche) following the manufacturer’s protocol. The sequencing was performed on NovaSeq 6000 (Illumina) with a read length of 2x50bp.
RNAseq data processing and analysis
RNAseq reads were mapped against the human reference genome (GRCh38) with STAR/2.7.8a using ENCODE parameters. Genes were quantified with RSEM/1.3.0 with default parameters using gencode.v42. Only genes with >1 cpm in at least 12 samples were retained for analysis. Differential expression was performed with limma84 using the voom85 transformation of the counts. Genes with FDR < 5% were considered significant. Functional enrichment was performed with g:profiler86 and gene set enrichment analysis with fgsea87 with a pre-ranked list of genes by the limma moderated t statistic.
Fibroblasts proliferation assay
Fibroblasts proliferation assays were performed using the Click-iT EdU Imaging Kit (Thermo Fisher Scientific, C10499), according to the manufacturer’s instructions. Briefly, for acute treatments (24 h) with CBD/vehicle, cells were plated at a density of 5 × 104 cells/mL. Six hours before fixation, permeabilization, and 5′-ethynyl-2′-deoxyuridine (EdU) staining, cells were incubated with EdU solution at a 10 μM final concentration. For chronic treatments (72 h) with CBD 5 µM (n = 3 independent cultures), CBD 0.5 µM (n = 4), CBD 0.1 µM (n = 6), or vehicle (n = 6), cells were seeded at a density of 1 × 104 cells/mL and incubated for 17 h with EdU solution (10 µM). EdU incorporation was finally quantified by using a fluorescence microplate reader using an excitation of 568 nm and an emission of 585 nm.
Measurement of ROS levels
Fibroblasts from LS patients bearing NDUFS4 or COX15 mutations and controls were seeded at ~35,000 cells/well on 12 mm glass coverslips and treated with 1 μM CBD or vehicle for 24 h. Cells were then washed three times with pre-warmed phosphate-buffered saline (PBS) and incubated for 20 min in HBSS buffer (Gibco™, #14025050) supplemented with 3 µM MitoSOX™ Red (ThermoFisher, #M36008) and 200 nM MitoTracker™ Deep Red FM (ThermoFisher, #M46753). Finally, cells were washed three times with PBS, fixed with 4% (v/v) paraformaldehyde for 15 min and mounted with Dapi-Fluoromount-G™ mounting medium (EMS, #17984-24). Images were acquired using confocal microscopy (LSM780, Zeiss, Germany) with the Cy3 and Cy5 channels. The red fluorescence intensity of each cell identified with full DAPI staining was analyzed using ImageJ software. A minimum of 200 cells were analyzed per condition. Three biological replicates were performed including three technical replicates in each. Rotenone (Santa Cruz #203242) (50 μM) was used as a positive control.
Measurement of total glutathione
Patient fibroblasts were cultured in Opti‐MEM medium lacking FBS for 72 h in order to avoid influence of the GSH contained in the FBS and treated with 1 µM CBD or vehicle for 24 h. Total GSH was measured by absorbance at 412 nm and adapted to 96-well plates with slight modifications88. Briefly, cell pellets were resuspended in ice‐cold phosphate buffer (50 mM sodium phosphate, 6.3 mM EDTA-Na2, pH 7.4), centrifuged at 16,000 × g for 30 min and the supernatant was used for biochemical analysis. For total glutathione determination, 20 µl of each sample was mixed with 180 µl of phosphate buffer containing 1 mM DTNB 20 U/ml glutathione reductase and 1 mM NADPH. The kinetics of the formation of TNB (5-thio-2-nitrobenzoic acid) were immediately recorded at 412 nm, every 30 s over a 2-min period. Glutathione concentrations were calculated according to the standard curve prepared and expressed as µmol/µg protein.
Western blot
Whole-cell lysates from cell culture and olfactory bulb tissue were obtained by sonication in a 2% (w/v) SDS sample buffer with 1X protease inhibitor cocktail (Sigma-Aldrich #P8340). Mitochondria were immunoprecipitated from olfactory bulb tissue via MACS Mitochondria Isolation kit (Miltenyi Biotech, #130-097-340) and OctoMACS gentle homogenizer (#130-096-427) according to manufacturer instructions89. An aliquot of the lysate was taken before incubation with anti-TOM22 magnetic beads and served as input fraction for verifying mitochondrial enrichment. Protein concentration was measured with the BCA protein assay kit (ThermoFisher). Thereafter, 30 µg of protein lysates were heat-denatured in Laemmli sample buffer (Bio-Rad Laboratories, Inc), subjected to 4–20% gradient SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc). Membranes were then blocked for 1 h with 5% (w/v) dried skimmed milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) and incubated overnight at 4 °C with primary antibodies (1:1,000 unless stated otherwise) against NDUFS4 (Abcam, #ab87399), COX5a (Abcam, #ab110262), PPARγ (Cell Signaling, #2435), CB1R (Frontier Bio, #CB1-GP-Af530), VDAC1 (Abcam, #ab154856, 1:2,000), TOM20 (Santa Cruz, #sc-11415), phospho-ERK1/2 (Cell Signaling, #9101), ERK1/2 (Cell Signaling, #9102), phospho-AKT1 (Cell Signaling, #9271), AKT1 (Cell Signaling, #2967), phospho-RPS6 (Cell Signaling, #4858), RPS6 (Cell Signaling, #2217) and GAPDH (GeneTex, #GTX627408, 1:20,000). After incubation with the corresponding HRP-conjugated secondary antibodies (1:10,000; Jackson Immuno Research), membranes were washed in TBS-T and developed using an enhanced chemiluminescence (ECL) detection system (Pierce). Bands were quantified using Image J software (National Institutes of Health, USA).
Immunofluorescence
Brain tissue preparation and immunofluorescence were performed as described90. Briefly, 30-μm thick sections were cut with a vibratome (Leica, France). Free-floating sections were rinsed three times 10 min in phosphate-buffered saline (PBS, 0.1 M, pH 7.5). After 15 min incubation in 0.2 % (vol/vol) Triton X-100 in PBS, sections were rinsed in PBS again and blocked for 1 h in a 3% BSA in PBS solution. Sections were incubated 24 h at 4 °C in 1 % BSA, 0.15 % Triton X-100 with the following primary antibodies: IBA1 (Wako, #019-19741, 1:1,000), GFAP (Abcam, #ab4674, 1:3,000), and TMEM119 (Abcam, #ab209064, 1:300). Sections were rinsed three times for 10 min in PBS and incubated for 45–60 min with goat Cy2-, Cy3- and Cy5-coupled (1:750, Jackson Immunoresearch) and/or goat Alexa Fluor 488 (1:750, Life Technologies). Sections were rinsed for 10 min twice in PBS and twice in phosphate-buffer (0.1 M, pH 7.5) before mounting in DAPI-Fluoromount-G solution (Electron Microscopy Sciences, USA). Images were acquired using confocal microscopy (LSM780, Zeiss) as described90. For each mouse, intensity signal quantification was performed delimiting the GPe in 4 coronal sections along the rostrocaudal axis within a window between −0.10 and −0.94 mm from bregma91. Images were analyzed using the gray-value measuring tool in NIH ImageJ. The intensity of each section was normalized by the surface area of the GPe. Data were quantified by averaging the normalized pixel intensity of 4 coronal sections/mouse.
Image acquisition and 3D morphometric microglia analyses with Imaris
Confocal images from IBA1 immunofluorescence were acquired using the z-stack function (~20 μm z-stack at 0.8 μm interval) of a LSM780 confocal microscope (Zeiss, Germany) with a 40x PlanApo oil-immersion objective at a resolution of 1024 × 1024 pixels. Microglia were reconstructed in 3D using Imaris (version 9.6, Bitplane, UK). To analyze cell morphology, the microglial surface was recreated with the following custom parameters: surface detail of 0.160 μm; diameter of largest sphere which fits into the object: 1.17; color: object ID. Additionally, a separated surface rendering was generated to manually reconstruct the soma volume of the microglia. Then, microglial cells with cytoplasm located in the middle of the stack and far from the tissue edge were specifically chosen for further analyses. The previously reconstructed surface served as a guide model for filament reconstruction using the following custom parameters: seed points 0.469 μm; detect new starting points: largest diameter 13 μm for cKO-Veh group and 7.81 μm for CT-Veh, CT-CBD and cKO-CBD groups. Given the differences found in soma sizes, groups were analyzed with different largest diameters values. Any inaccuracies in the initial placement of dendrite beginning points by the Imaris algorithm were manually corrected. If the algorithm placed the points incorrectly, they were adjusted by either placing them accurately at the center of the soma or removing them if necessary. Furthermore, in cases where the software mis-identified fused microglial cells as a single entity, they were either manually separated by selecting a specific point and using the delete function, or completely removed from the sample if separation was not possible. Finally, dendrite volume and dendrite branch level from Imaris’ Filament Tracer were subjected to detailed analysis. Both the 3D reconstruction and data analysis were conducted in a blind manner regarding the experimental conditions.
Tagged-ribosome immunoprecipitation and RNA extraction
HA-tagged-ribosome immunoprecipitation was performed as described90. Briefly, the olfactory bulb and cerebellum of Gad2:Ndufs4CT-RiboTag and Gad2:Ndufs4cKO-RiboTag mice were homogenized by douncing in 1-ml polysome buffer (50 mM Tris, pH 7.4, 100 mM KCl, 12 mM MgCl2, and 1 % NP-40 supplemented with 1 mM DTT, 1 mg/ml heparin, 100 μg/ml cycloheximide, 200 U/ml RNAseOUT, and protease inhibitor mixture). Samples were then centrifuged at 10,000 × g for 10 min to collect the postmitochondrial supernatant. Then, 100 μl of each supernatant was transferred to a new tube serving as input fraction. Anti-HA antibody (5 μl/sample; Covance, #MMS-101R) was added to the remaining supernatant and incubated overnight at 4 °C with constant gently rotation. The following day, samples were added to protein G magnetic beads (Invitrogen, #100.04D) and incubated overnight at 4 °C with constant gently rotation. On the third day, magnetic beads were washed twice in a magnetic rack for 10 min each in high-salt buffer (50 mM Tris, pH 7.4, 300 mM KCl, 12 mM MgCl2, 1 % NP-40, 1 mM DTT, and 100 μg/ml cycloheximide). After washing, 350 μl of Qiagen RLT buffer (supplemented with β-Mercaptoethanol) were added to the pellets and to the input samples. Total RNA was extracted from ribosome-mRNA complexes as well as from the input fraction (total homogenate) using a RNeasy Microkit (Qiagen). Quality and quantity of RNA samples were both assessed using an Agilent Bioanalyzer 2100 system (Agilent Technologies).
Quantitative real-time PCR
Total cellular RNA was extracted using the RNeasy Mini Kit (Qiagen, #74106) according to manufacturer’s instructions, treated with DNAse (RNase-Free DNase Set, Qiagen, #79254) and subsequently reversely transcribed with the TaqMan RNA-to-Ct 1-step kit (ThermoFisher, #4392938). Amplification was performed by quantitative real-time PCR, using the standard curve method with specific Taqman probes (from Custom TaqMan Gene Expression Assay, ThermoFisher Scientific) for the targeted human gene PPARG (Hs01115513_m1), mouse Pparg (Mm01184323_m1), mouse Cnr1 (Mm01212171_s1), mouse Cnr2 (Mm00438286_m1), mouse Gpr55 (Mm02621622_s1), mouse Mt1 (Mm00496660_g1), mouse Mt2 (Mm00809556_s1), mouse Slc3a2 (Mm00500521_m1), mouse Chst2 (Mm00490018_g1) and the respective human/mouse GAPDH/Gapdh probes as housekeeping control genes (Hs02786624_g1/ Mm99999915_g1).
Hippocampal slices preparation
Male and female mice at PND40-50 were deeply anesthetized with isoflurane and killed by decapitation. The brain was removed and quickly placed in an ice-cold cutting solution containing 200 mM sucrose, 26 mM NaHCO3, 10 mM glucose, 3 mM KCl, 1.25 mM NaH2PO4, 4 mM MgSO4, and 0.5 mM CaCl2. Coronal hippocampal slices (300 µm thick) were prepared with a Leica VT1000S vibratome. After sectioning, slices were placed in a holding chamber containing standard artificial cerebrospinal fluid solution (ACFS): 124 mM NaCl, 26 mM NaHCO3, 10 mM glucose, 3 mM KCl, 1.25 mM NaH2PO4, 1 mM MgSO4, and 2 mM CaCl2, and then stored at 22–24 °C. After an incubation period of 1 h, slices were transferred to a submersion recording chamber at RT. All incubation solutions were continuously bubbled with a gas mixture of 95% O2/5% CO2.
Ex-vivo electrophysiology
The recording chamber was perfused with ACSF gassed with 5% CO2/95% O2. Electrophysiological recordings were performed using a Multiclamp 700B amplifier (Molecular Devices). Synaptic responses were evoked using single-voltage pulses (up to 10 mA) delivered through an ISO-Flex stimulator (A.M.P.I) connected to a patch micropipette filled with ACSF. The stimulating electrodes were placed over the Schaffer collateral fibers in the stratum radiatum area. Field excitatory postsynaptic potentials (fEPSPs) were recorded with low-resistance glass electrodes filled with ACSF placed in the apical dendritic layer of CA1 area. Baseline stimulation was delivered every 15 s (0.01 ms pulses) for 8–10 min first in regular ACSF and later in the presence of picrotoxin 100 µM. A second set of experiments was performed in the same conditions after a minimum of 1 h incubation in the presence of CBD 16 µM.
Electrophysiological in vivo data acquisition
For all experiments, data acquisition was performed as described23. Briefly, local field potential activity was amplified, A-D converted and sampled at 1 KHz and bandpass filtered at 0.1–250 Hz (DigitalLynx SX and Cheetah Data Acquisition System, Neuralynx, USA). Continuous spike signals were also recorded, amplified, band-pass filtered (300 Hz to 8 KHz) and sampled at 32 KHz.
Electrophysiological in vivo data analysis
For the presence of seizures, data were analyzed in Spike2 (Cambridge Electronic Design Limited, UK) for visual inspection and report presence of epileptic events. Offline single-unit spike sorting was performed with Offline Sorter software (Offline Sorter, Plexon Inc, USA). Briefly, for each channel, a specific manual threshold was defined and all events bypassing this threshold were assumed to be an action potential. After an overall waveform shape and three principal component analysis (PCA) inspection, all spikes were sorted with an automatic K-mean algorithm to separate clusters of cells.
Surgery and electrodes placements
Gad2:Ndufs4cKO and control mice were first injected with analgesic (ketoprofen 5 mg/kg, s.c.) to control pain, and then induced and anesthetized with a mixture of isoflurane and O2 (2–5% isoflurane induction, 1.5% maintenance at 0.4–1.0 L/min O2 flow). During the surgery, the body core temperature (37 °C) and physiological states (breathing, vibrissae movement) were continuously monitored. Animals were placed in a Robot Stereotaxic (Neurostar, Germany) and the scalp was incised and small burr holes were made in the skull above the motor cortex on both hemispheres under stereotaxic guidance according to Paxinos91 (from bregma in mm, AP = +1.50, ML = +/−2.00). Each electrocorticogram ECoG electrode consisted of a micro-screw attached to a fine diameter silver wire (130 mm bare; 180 mm coated; Advent Research Material Ltd., England). The screw electrodes were placed through the small cranial burr holes at visually identified locations. A reference electrode was placed at the midline cerebellum and a ground electrode was placed subcutaneously over the back and the skin was closed with sutures. All electrodes were previously soldered to an MilMax connector (Mill-Max 2 × 9 MM Male-to-Male Header, Neuralynx, USA). After surgery, animal welfare and body weight were documented daily for 3 days and mice were allowed to recover for 14 days before any experimentation.
In vivo ECoG recordings and thermal seizure induction
Recording approach was adapted from23. Depending on the experiment, CBD (100 mg/kg, i.p.) or vehicle injection were performed 1 h before the thermal seizure induction or right after seizure onset. Recordings consist of simultaneous video-ECoG-records collected from freely moving mice in their home cage after being connected to a headstage linked to a Saturn Commutator (Neuralynx, USA). ECoG activity was amplified, A-D converted and sampled at 1KHz and bandpass filtered at 0.1 to 250 Hz (DigitalLynx SX and Cheetah Data Acquisition System, Neuralynx, USA). An infrared heat lamp (HL-1, Phymep, France) was table mounted with an adjustable swing arm for easy adjustment of radiant heat to specimen. After 10–15 min of basal activity recording, the lamp was switched on and mouse body temperature was controlled every 20 min using a rectal temperature probe (RET-4, Phymep, France) connected to a temperature controller (CMA 450 Temperature controller, Phymep, France). When tonic-clonic seizures started, the lamp was immediately switched off and the animal’s temperature was reported. Electrophysiological activity and mice welfare and behavior was recorded for 1 h minimum after seizure onset.
Mitochondria isolation and respiration assays
Mitochondria from control and Ndufs4KO tissues were isolated as described92. Briefly, mice at their late stage of the disease were sacrificed 30 min after the treatment with CBD (100 mg/kg/day) or vehicle and the olfactory bulbs were extracted. A similar procedure was performed in mice exposed to leriglitazone (50 mg/kg/day) or regular food, as well as CB1R-KO mice and wild-type littermates from which hippocampal tissue was used instead. The olfactory bulbs were dissected and homogenized using a glass dounce homogenizer containing MAS (Mitochondrial Assay Solution) buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2 mM HEPES-KOH, pH 7.2). Homogenates were centrifuged at 900 × g for 10 min at 4 °C. Supernatants were further centrifuged at 9000 × g for 10 min at 4 °C. Resulting supernatants were used for L-lactate measurements (Biosystems) and pellets containing mitochondria were resuspended in MAS buffer. Protein concentration was determined using the BCA protein assay (ThermoFisher Scientific) according to the manufacturer’s instructions. Oxygen consumption rate (OCR) was measured in MAS buffer supplemented with 0.5% (w/v) fatty acid–free BSA (MAS-BSA), using a Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies, Inc). Freshly isolated mitochondria (10–25 μg of protein) were plated in each well of an XFp plate in 25 μL of MAS-BSA buffer and centrifuged at 2000 × g at 4 °C for 20 min. Subsequently, 155 μL of MAS-BSA buffer containing 10 mM pyruvate/5 mM malate was added to each well. First, basal measurements of oxygen consumption rate (state II) were obtained. Next, ADP (5 mM) was injected (state III), followed by sequential injections of oligomycin (2 μM, state IV), FCCP (5 μM, state IIIu) and antimycin A/rotenone (2.5 μM and 2 μM, respectively) to disrupt mitochondrial respiration. Together, this injection series allowed for the determination of ADP-uncoupled respiration (ADP), proton leak (oligomycin), maximal respiration (FCCP) and non-mitochondrial residual oxygen consumption (antimycin A/rotenone). For hippocampal mitochondria, analyses were carried out using a 2 K Oroboros device93. 25 μl of lysate were put in each chamber and complex I-dependent respiration was triggered by adding malate (2 mM), pyruvate (5 mM) and glutamate (10 mM) (MPG)93. Then we applied DMSO, CBD or WIN 55,212-2 and 5 min after we injected 1.25 mM ADP. Each measure of OCR in ADP condition was normalized to the values before ADP injection and the effect of WIN 55,212-2 and CBD was expressed as percentage of vehicle conditions. Similarly, the measure of succinate condition was normalized to ADP vehicle. Only samples for which the ratio of ADP/MPG in the vehicle was equal or superior to 1.5 were retained for the analyses.
Blue native in gel activity assay
Isolated mitochondria from olfactory bulbs of late-stage Ndufs4KO and control mice treated with either vehicle or CBD (100 mg/kg/day) were isolated as described94. After being resuspended in 1.5 M aminocaproic acid, 50 mM Bis-Tris-HCl (pH 7) and 4 mg digitonin per milligram of protein, mitochondria were incubated for 5 min on ice and subsequently centrifuged at 20,000 × g for 30 min at 4 °C. 5% (w/v) Coomassie G250 was then added to the supernatant. 50–100 μg were separated by 4–20% gradient SDS-PAGE blue native gel electrophoresis and either stained for in-gel activities (CI and CIV) or electroblotted on PVDF membrane for immunodetection with specific antibodies against CI (NDUFS3, Abcam, #ab110246) and CII (SDHB, Abcam, #ab14714).
Primary neuronal cell culture
Brainstem neurons from PND4-7 Ndufs4KO mice and WT littermates were cultured as described95 with some modifications. Briefly, brainstems were dissected from the whole brain and blood vessels, meninges, and the cerebellum were carefully removed. Brainstems were enzymatically dissociated with papain followed by mechanical dissociation by pipetting in and out. Dissociated cells were plated in Neurobasal-A (NBA, Thermo Fisher; 10888022) medium supplemented with B-27 and 1% FBS in laminin and polylysine coated plates. At day in vitro (DIV) 4, fresh supplemented NBA plus AraC 4.8 µM; MERCK; C6645) was added to the cultures to stop glial proliferation. Neurons were cultured for a period of 10-11 days when experiments were performed.
For astrocyte cultures, cortical homogenates of PND4-7 mice were grown in DMEM, 4.5 g/L glucose supplemented with 1% (v/v) glutamine, and 10% (v/v) FBS. After approximately 10 days, astrocytic cultures were agitated on a mechanical shaker at 400 rpm for 4 h to eliminate superficial microglia, washed twice with PBS, detached with trypsin/EDTA, and re-seeded.
Plasmids, transfections, and luciferase reporter assay
PPAR-Luc plasmids were made by removing 3XDR1 sites with MluI and HindIII from the PPREX3-TK-Luc plasmid, a gift from Bruce Spiegelman (Addgene plasmid #1015; http://n2t.net/addgene:1015; RRID:Addgene_1015), and subcloning it into a minimal fos promoter-driven luciferase reporter vector obtained by removing ATF6 binding sites in the plasmid p5xATF6-GL3, a gift from Ron Prywes (Addgene plasmid #11976; http://n2t.net/addgene:11976; RRID:Addgene_11976), with XhoI and HindIII.
pGL4.73 [hRLuc/SV40] (Promega) plasmid was digested with HindIII and XbaI to obtain Renilla (RLuc) coding sequence. RLuc was cloned in pEF1 plasmid (Invitrogen) in EcoRV and XbaI sites.
Transfections were performed with Lipofectamine 2000 (Thermo Scientific). Prior to transfection, the neurons were transferred from NBA medium to a TMits medium, composed of 10% MEM (Invitrogen) and 90% salt-glucose-glycine (SGG) medium (SGG: 114 mM NaCl, 0.2% NaHCO3, 5.3 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM glucose, 1 mM glutamine, 0.5 mM sodium pyruvate, 0.1% phenol red; osmolarity 325 mOsm/L). Astrocytes were transfected in OptiMEM. Twenty-four hours after transfection, cells were treated with CBD or vehicle for additional 24 h before luciferase assays were performed using the Dual Glo Luciferase Assay system (Promega) with firefly luciferase-based reporter gene activity normalized to the RLuc control.
Behavioral assays
Mice were handled for 3 days prior to testing for habituation. Since there was no evidence of sex differences in our behavioral measurements, data from male and female mice were pooled. All experiments were conducted in a blind manner regarding genotype and treatment.
Evaluation of clinical signs
Clasping and abnormal trunk twisting behaviors were used as signs of neurological degeneration, as described82. Clasping is defined as an inward curling of the spine and a retraction of the forelimbs and/or hindlimbs toward the midline of the body.
Rotarod
Rotarod was performed at PND30, 40, and 50. Mice were placed on a rotating drum that gradually accelerated from 4 to 40 rpm over 300 s. Each trial ended when a mouse fell off, made one complete backward revolution while hanging on, or reached 5-min. Each mouse was tested in 3 trials/day spaced 20 min apart. The results of the 3 trials were averaged.
Whole-body plethysmography
Respiratory frequency, tidal volume (normalized per body weight), and total ventilation were assessed under normoxic conditions using whole-body, flow plethysmography (EMMS, UK) in unanesthetized, freely moving PND43-45 Ndufs4KO and control mice as described23.
Three-chamber social approach
A three-chamber arena was used to assess sociability in PND48-55 Gad2:Nudfs4cKO and control mice as described96. Briefly, on day 1, a stranger target mouse was habituated to the wire cups. On day 2, a test mouse was placed in the middle chamber and allowed to freely explore all the empty chambers of the apparatus for 10 min. Next, an unfamiliar mouse (age and gender matched) was introduced into one of the two side chambers, enclosed in a wire cage allowing only for the test mouse to initiate any social interaction. An identical empty wire cage was placed in the other side chamber. Following placement, the test mouse was allowed to explore the whole three-chamber arena for 10 min. The time spent sniffing the unfamiliar mouse and the empty wire cage was manually scored. The discrimination index for sociability was calculated as follows: (time exploring unfamiliar mouse—time exploring empty wire cage)/(total exploration time) * 100. Mice with exploration times <10 s for either the object or the stranger were excluded.
Elevated plus maze
The elevated plus maze was performed in Gad2:Nudfs4cKO and control mice at PND48-55 as described97. The maze consisted of a Plexiglas apparatus with four arms (29 cm long × 5 cm wide) arranged in a cross shape extending from a neutral central square (5 cm × 5 cm), elevated 40 cm above the floor. Five-minute test sessions were performed. The percentage of time spent in the open arms and the number of total arm entries were recorded.
Statistical analyses
GraphPad Prism v9.0 software was used for statistical analyses. Data are shown as the mean ± SEM. All statistical analyses were performed using either one-way analysis of variance (ANOVA) for multiple comparisons followed by Tukey’s or Dunnett’s post hoc tests, two-, or three-way ANOVA followed by Tukey’s post hoc test. Student’s t test (unpaired or paired, two-tailed) was used for comparison between two groups, when relevant. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. All datasets were analyzed to fit a normal model by Kolmogorov–Smirnoff test and met the core assumptions to validate application of the abovementioned statistical tests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA Sequence data generated in this study have been deposited in Gene Expression Omnibus, under the accession code GSE266113. The data supporting the findings of this study and the data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Friedman, J. R. & Nunnari, J. Mitochondrial form and function. Nature 505, 335–343 (2014).
Vafai, S. B. & Mootha, V. K. Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383 (2012).
Viscomi, C. & Zeviani, M. Experimental therapy for mitochondrial diseases. Handb. Clin. Neurol. 194, 259–277 (2023).
Arii, J. & Tanabe, Y. Leigh syndrome: serial MR imaging and clinical follow-up. AJNR Am. J. Neuroradiol. 21, 1502–1509 (2000).
Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr. Neurol. 39, 223–235 (2008).
Lee, H.-F., Tsai, C.-R., Chi, C.-S., Lee, H.-J. & Chen, C. C.-C. Leigh syndrome: clinical and neuroimaging follow-up. Pediatr. Neurol. 40, 88–93 (2009).
Lake, N. J., Compton, A. G., Rahman, S. & Thorburn, D. R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 79, 190–203 (2016).
Kruse, S. E. et al. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 7, 312–320 (2008).
Quintana, A. et al. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J. Clin. Investig. 122, 2359–2368 (2012).
van de Wal, M. et al. Ndufs4 knockout mouse models of Leigh syndrome: pathophysiology and intervention. Brain, 145, 45–63 (2021).
Debray, F.-G. et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 119, 722–733 (2007).
Pfeffer, G., Majamaa, K., Turnbull, D. M., Thorburn, D. & Chinnery, P. F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, CD004426 (2012).
Pfeffer, G. et al. New treatments for mitochondrial disease-no time to drop our standards. Nat. Rev. Neurol. 9, 474–481 (2013).
Marsicano, G., Moosmann, B., Hermann, H., Lutz, B. & Behl, C. Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J. Neurochem. 80, 448–456 (2002).
Bhunia, S., Kolishetti, N., Arias, A. Y., Vashist, A. & Nair, M. Cannabidiol for neurodegenerative disorders: A comprehensive review. Front Pharm. 13, 989717 (2022).
Schon, E. A. & Przedborski, S. Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053 (2011).
Mechoulam, R., Peters, M., Murillo-Rodriguez, E. & Hanus, L. O. Cannabidiol-recent advances. Chem. Biodivers. 4, 1678–1692 (2007).
Izzo, A. A., Borrelli, F., Capasso, R., Di Marzo, V. & Mechoulam, R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 30, 515–527 (2009).
Ryan, D., Drysdale, A. J., Lafourcade, C., Pertwee, R. G. & Platt, B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 29, 2053–2063 (2009).
Nichols, J. M. & Kaplan, B. L. F. Immune Responses Regulated by Cannabidiol. Cannabis Cannabinoid Res 5, 12–31 (2020).
Silvinato, A., Floriano, I. & Bernardo, W. M. Use of cannabidiol in the treatment of epilepsy: Lennox-Gastaut syndrome, Dravet syndrome, and tuberous sclerosis complex. Rev. Assoc. Med Bras. 68, 1345–1357 (2022). (1992).
Johnson, S. C. et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528 (2013).
Bolea, I. et al. Defined neuronal populations drive fatal phenotype in a mouse model of Leigh syndrome. Elife 8, e47163 (2019).
Haas, R. H. Autism and mitochondrial disease. Dev. Disabil. Res Rev. 16, 144–153 (2010).
Manning, A. et al. Elevated susceptibility to exogenous seizure triggers and impaired interneuron excitability in a mouse model of Leigh syndrome epilepsy. Neurobiol. Dis. 187, 106288 (2023).
Deiana, S. et al. Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Δ9-tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive-compulsive behaviour. Psychopharmacology 219, 859–873 (2012).
Ohlsson, A. et al. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed. Environ. Mass Spectrom. 13, 77–83 (1986).
Kaplan, J. S., Stella, N., Catterall, W. A. & Westenbroek, R. E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl Acad. Sci. USA 114, 11229–11234 (2017).
Lim, A. & Thomas, R. H. The mitochondrial epilepsies. Eur. J. Paediatr. Neurol. 24, 47–52 (2020).
Sutton, F. et al. Testing blood and CSF in people with epilepsy: a practical guide. Epileptic Disord. 22, 381–398 (2020).
Cuesta, P. et al. Gamma band functional connectivity reduction in patients with amnestic mild cognitive impairment and epileptiform activity. Brain Commun. 4, fcac012 (2022).
Song, H. et al. Contributions of the Hippocampal CA3 Circuitry to Acute Seizures and Hyperexcitability Responses in Mouse Models of Brain Ischemia. Front. Cell Neurosci. 12, 278 (2018).
Jensen, K. R., Berthoux, C., Nasrallah, K. & Castillo, P. E. Multiple cannabinoid signaling cascades powerfully suppress recurrent excitation in the hippocampus. Proc. Natl Acad. Sci. USA 118, e2017590118 (2021).
Rosenberg, E. C. et al. Cannabidiol modulates excitatory-inhibitory ratio to counter hippocampal hyperactivity. Neuron 111, 1282–1300.e1288 (2023).
Martin-Perez, M. et al. PKC downregulation upon rapamycin treatment attenuates mitochondrial disease. Nat. Metab. 2, 1472–1481 (2020).
Stokes, J. C. et al. Leukocytes mediate disease pathogenesis in the Ndufs4(KO) mouse model of Leigh syndrome. JCI Insight 7, e156522 (2022).
Peng, J. et al. A narrative review of molecular mechanism and therapeutic effect of cannabidiol (CBD). Basic Clin. Pharm. Toxicol. 130, 439–456 (2022).
Bennett, M. L. et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl Acad. Sci. USA 113, E1738–E1746 (2016).
Calvaruso, M. A. et al. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet 21, 115–120 (2012).
Kouchaeknejad, A., Van Der Walt, G., De Donato, M. H. & Puighermanal, E. Imaging and Genetic Tools for the Investigation of the Endocannabinoid System in the CNS. Int. J. Mol. Sci. 24, 15829 (2023).
Aguirre-Rueda, D. et al. WIN 55,212-2, agonist of cannabinoid receptors, prevents amyloid β1-42 effects on astrocytes in primary culture. PLoS ONE 10, e0122843 (2015).
Marsicano, G. et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534 (2002).
Carlos, N.-O. & Guillermo, A. Y. The Multiple Waves of Cannabinoid 1 Receptor Signaling. Mol. Pharm. 90, 620 (2016).
Biever, A., Valjent, E. & Puighermanal, E. Ribosomal Protein S6 Phosphorylation in the Nervous System: From Regulation to Function. Front. Mol. Neurosci. 8, 75 (2015).
Puighermanal, E. et al. Dissociation of the pharmacological effects of THC by mTOR blockade. Neuropsychopharmacology 38, 1334–1343 (2013).
Corona, J. C. & Duchen, M. R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 100, 153–163 (2016).
Costa, A.-M. et al. Antiseizure Effects of Cannabidiol Leading to Increased Peroxisome Proliferator-Activated Receptor Gamma Levels in the Hippocampal CA3 Subfield of Epileptic Rats. Pharmaceuticals 15, 495 (2022).
Rodríguez-Pascau, L. et al. The brain penetrant PPARγ agonist leriglitazone restores multiple altered pathways in models of X-linked adrenoleukodystrophy. Sci. Transl. Med. 13, eabc0555 (2021).
Rodríguez-Pascau, L. et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol. Dis. 148, 105162 (2021).
Sanz, E. et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl Acad. Sci. USA 106, 13939–13944 (2009).
González-Quintana, A. et al. Uniparental isodisomy as a cause of mitochondrial complex I respiratory chain disorder due to a novel splicing NDUFS4 mutation. Mol. Genet Metab. 131, 341–348 (2020).
Juárez-Rebollar, D., Rios, C., Nava-Ruíz, C. & Méndez-Armenta, M. Metallothionein in Brain Disorders. Oxid. Med Cell Longev. 2017, 5828056 (2017).
Liu, J., Xia, X. & Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 28, 2358–2366 (2020).
Katila, N., Bhurtel, S., Park, P.-H. & Choi, D.-Y. Metformin attenuates rotenone-induced oxidative stress and mitochondrial damage via the AKT/Nrf2 pathway. Neurochem. Int. 148, 105120 (2021).
Juknat, A. et al. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglial cells. Br. J. Pharmacol. 165, 2512–2528 (2012).
Jain, I. H. et al. Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61 (2016).
Ferrari, M. et al. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl Acad. Sci. USA 114, E4241–E4250 (2017).
Perry, E. A. et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 3, 33–42 (2021).
von Wrede, R., Helmstaedter, C. & Surges, R. Cannabidiol in the Treatment of Epilepsy. Clin. Drug Investig. 41, 211–220 (2021).
Lee, S., Na, J.-H. & Lee, Y.-M. Epilepsy in Leigh Syndrome With Mitochondrial DNA Mutations. Front. Neurol. 10, 496 (2019).
Rahman, S. Mitochondrial disease and epilepsy. Dev. Med Child Neurol. 54, 397–406 (2012).
Gu, B. et al. Cannabidiol attenuates seizures and EEG abnormalities in Angelman syndrome model mice. J. Clin. Investig. 129, 5462–5467 (2019).
Aguilar, K. et al. Microglial response promotes neurodegeneration in the Ndufs4 KO mouse model of Leigh syndrome. Glia 70, 2032–2044 (2022).
Hampson, A. J., Grimaldi, M., Axelrod, J. & Wink, D. Cannabidiol and (−)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl Acad. Sci. USA 95, 8268–8273 (1998).
Miller, H. C. et al. Metallothionein 1 Overexpression Does Not Protect Against Mitochondrial Disease Pathology in Ndufs4 Knockout Mice. Mol. Neurobiol. 58, 243–262 (2021).
Nuhu, F., Gordon, A., Sturmey, R., Seymour, A.-M. & Bhandari, S. Measurement of Glutathione as a Tool for Oxidative Stress Studies by High Performance Liquid Chromatography. Molecules 25, 4196 (2020).
Yoon, J.-Y. et al. Metabolic rescue ameliorates mitochondrial encephalo-cardiomyopathy in murine and human iPSC models of Leigh syndrome. Clin. Transl. Med. 12, e954 (2022).
Iuso, A. et al. Dysfunctions of Cellular Oxidative Metabolism in Patients with Mutations in the NDUFS1 and NDUFS4 Genes of Complex I *. J. Biol. Chem. 281, 10374–10380 (2006).
van Rensburg, D. J., Lindeque, Z., Harvey, B. H. & Steyn, S. F. Ndufs4 KO mice: A model to study comorbid mood disorders associated with mitochondrial dysfunction. Pharmacol. Biochem. Behav. 234, 173689 (2024).
Russell, O. M., Gorman, G. S., Lightowlers, R. N. & Turnbull, D. M. Mitochondrial Diseases: Hope for the Future. Cell 181, 168–188 (2020).
Maresz, K., Carrier, E. J., Ponomarev, E. D., Hillard, C. J. & Dittel, B. N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 95, 437–445 (2005).
Rosen, C. J. Revisiting the rosiglitazone story-lessons learned. N. Engl. J. Med. 363, 803–806 (2010).
Pinto, M. et al. Pioglitazone ameliorates the phenotype of a novel Parkinson’s disease mouse model by reducing neuroinflammation. Mol. Neurodegeneration 11, 25 (2016).
Lebovitz, H. E. Thiazolidinediones: the Forgotten Diabetes Medications. Curr. Diab. Rep. 19, 151 (2019).
Chang, R. C. et al. Cannabidiol Promotes Adipogenesis of Human and Mouse Mesenchymal Stem Cells via PPARγ by Inducing Lipogenesis but Not Lipolysis. Biochem Pharm. 197, 114910 (2022).
Senn, L. et al. Is the peroxisome proliferator-activated receptor gamma a putative target for epilepsy treatment? Current evidence and future perspectives. Pharmacol. Therapeutics 241, 108316 (2023).
Simeone, T. A., Matthews, S. A., Samson, K. K. & Simeone, K. A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 287, 54–64 (2017).
O’Sullivan, S. E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 173, 1899–1910 (2016).
Khosropoor, S., Alavi, M. S., Etemad, L. & Roohbakhsh, A. Cannabidiol goes nuclear: The role of PPARγ. Phytomedicine 114, 154771 (2023).
Lyu, J. et al. Bezafibrate Rescues Mitochondrial Encephalopathy in Mice via Induction of Daily Torpor and Hypometabolic State. Neurotherapeutics 19, 994–1006 (2022).
Pizcueta, P. et al. Development of PPARγ Agonists for the Treatment of Neuroinflammatory and Neurodegenerative Diseases: Leriglitazone as a Promising Candidate. Int. J. Mol. Sci. 24, 3201 (2023).
Quintana, A., Kruse, S. E., Kapur, R. P., Sanz, E. & Palmiter, R. D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl Acad. Sci. USA 107, 10996–11001 (2010).
Köhler, W. et al. Safety and efficacy of leriglitazone for preventing disease progression in men with adrenomyeloneuropathy (ADVANCE): a randomised, double-blind, multi-centre, placebo-controlled phase 2–3 trial. Lancet Neurol. 22, 127–136 (2023).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Law, C. W., Chen, Y., Shi, W., Smyth, G. K. & voom precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Korotkevich, G. et al. Fast gene set enrichment analysis, bioRxiv 060012; https://doi.org/10.1101/060012 (2021).
Rahman, I., Kode, A. & Biswas, S. K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 1, 3159–3165 (2006).
Minet, A. D. & Gaster, M. ATP synthesis is impaired in isolated mitochondria from myotubes established from type 2 diabetic subjects. Biochem. Biophys. Res. Commun. 402, 70–74 (2010).
Puighermanal, E. et al. Anatomical and molecular characterization of dopamine D1 receptor-expressing neurons of the mouse CA1 dorsal hippocampus. Brain Struct. Funct. 222, 1897–1911 (2017).
Paxinos, G. & Franklin, K. B. Paxinos and Franklin’s the mouse brain in stereotaxic coordinates, (Academic Press, 2019).
Gella, A. et al. Mitochondrial Proteome of Affected Glutamatergic Neurons in a Mouse Model of Leigh Syndrome. Front. Cell Dev. Biol. 8, 660 (2020).
Makrecka-Kuka, M., Krumschnabel, G. & Gnaiger, E. High-Resolution Respirometry for Simultaneous Measurement of Oxygen and Hydrogen Peroxide Fluxes in Permeabilized Cells, Tissue Homogenate and Isolated Mitochondria. Biomolecules 5, 1319–1338 (2015).
Fernández-Vizarra, E., López-Pérez, M. J. & Enriquez, J. A. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods 26, 292–297 (2002).
Kivell, B. M., McDonald, F. J. & Miller, J. H. Method for serum-free culture of late fetal and early postnatal rat brainstem neurons. Brain Res. Brain Res. Protoc. 6, 91–99 (2001).
Puighermanal, E. et al. Functional and molecular heterogeneity of D2R neurons along dorsal ventral axis in the striatum. Nat. Commun. 11, 1957 (2020).
Busquets-Garcia, A. et al. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol. Psychiatry 70, 479–486 (2011).
Acknowledgements
The authors thank all the lab members and Gabriel Mel de Fontenay for their feedback, the Microscopy Imaging Platform from Autonomous University of Barcelona for their help in the Imaris software, and Dr. Marta Gut for RNAseq. This work was supported by three MINECO Ramon y Cajal fellowships (RYC2020-029596-I; E.P., RYC2019-028501-I; E.S., and RyC-2012-11873; A.Q.), a Beatriu de Pinós fellowship from University and Research Grants Catalan Management Agency (#2017BP00132; E.P.) Marie Sklodowska-Curie Individual Fellowship (H2020-MSCA-IF-2014-658352; E.S., H2020-MSCA-IF-2020-101030783, M.L-S., H2020-MSCA-COFUND-2014-665919, A.G.), pre-doctoral fellowships (2018FI_B 00452 to AU, 2024 FI-1 00645 to GvdW). E.P received funds from MICINN (PID2021-125079OA-I00). E.S received funds from MICIU Proyectos I + D + i “Retos Investigacion” (RTI2018-101838-J-I00) and Ministerio de Ciencia e Innovación (MICINN) (PID2019-107633RB-I00 and PID2022-142544OB-I00). A.Q. is a recipient of an ICREA Academia award and received funds from the European Research Council (Starting grant NEUROMITO, ERC-2014-StG-638106), Grants SAF2014-57981P; SAF2017-88108-R, PID2020-114977RB-I00, RTC2019-006825-1, PDC2021-121883-I0, RED2022-134786-T, funded by MICIU/AEI /10.13039/5011000110330 and by MICIU/AEI /10.13039/501100011033 and Next GenerationEU/ PRTR, AGAUR (2017SGR-323, 2021SGR-720), Fundació TV3-La Marató (202030), and “la Caixa” Foundation (ID 100010434), under the agreement LCF/PR/HR20/52400018.
Author information
Authors and Affiliations
Contributions
E.P. and A.Q. designed the study and led the project. E.P., E.S, and A.Q. conceptualized the project. E.P. performed all behavioral studies related to CBD. E.P., I.A., P.B., and G.vW performed the behavioral studies with leriglitazone. M.L. performed qRT-PCR and in-gel-activity studies. M.L. and G.vW performed western blot analyses. A.G. performed mitochondrial assays. A.U., M.L. and P.B. performed fibroblast cultures assays. M.R. and S.J. performed in vitro electrophysiology studies. P.T. and FX.S. performed in vitro primary cultures. E.P. and F.M. performed thermal seizure induction. F.B. performed in vivo ECoG analyses. A.E-C, performed bioinformatic analyses of RNAseq. L.B. and G.M. performed OROBOROS mitochondrial assays. L.R.P., M.M., C.V, and P.P. provided leriglitazone and helped with experimental designs. E.P. and MH.dD. performed immunofluorescence experiments and quantification studies. MH.dD. performed IMARIS analyses. E.P., M.G-P., and M.L. performed ROS measurement experiments. E.P. and A.Q. wrote the manuscript with input from all authors. E.P., A.Q. and G.vW edited and proofread the manuscript between peer-review rounds with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
E.P., E.S., and A.Q. are listed as inventors on a patent application submitted by Autonomous University of Barcelona for the use of CBD described in this paper. L.R-P., P.P., C.V., and M.M. are current employees at Minoryx Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Carlo Viscomi, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Puighermanal, E., Luna-Sánchez, M., Gella, A. et al. Cannabidiol ameliorates mitochondrial disease via PPARγ activation in preclinical models. Nat Commun 15, 7730 (2024). https://doi.org/10.1038/s41467-024-51884-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51884-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
