recan [9] is a Python package which allows to construct genetic distance plots to explore and discover recombination events in viral genomes. This method has been previously implemented in desktop software tools: RAT[1], Simplot[2] and RDP4 [8].
Recan django-based web version is currently under development https://github.com/babinyurii/recan_gui
It is available at: http://yuriyb.pythonanywhere.com/
To use recan, you will need:
- Python 3
- Biopython
- plotly
- pandas
- Jupyter notebook
To install the package via pip run :
$ pip install recan
If you are going to use recan in JupyterLab, follow the insctructions to install the Jupyter Lab Plotly renderer
The package is intended to be used in Jupyter notebook.
Import Simgen class from the recan package:
from recan.simgen import Simgencreate an object of the Simgen class. To initialize the object pass your alignment in fasta format as an argument:
sim_obj = Simgen("./datasets/hbv_C_Bj_Ba.fasta")The input data are taken from the article by Sugauchi et al.(2002). This paper describes recombination event observed in hepatitis B virus isolates.
The object of the Simgen class has method get_info() which shows information about the alignment.
sim_obj.get_info()index: sequence id:
0 AB048704.1_genotype_C_
1 AB033555.1_Ba
2 AB010291.1_Bj
alignment length: 3215
We have three sequences in our alignment. Simgen class is based upon the MultipleSequenceAlignment class of the Biopython library. So, we treat our alignment as the array with n_samples and n_features, where 'samples' are sequences themselves, and the features are columns of nucleotides in the alignment. Index corresponds to the sequence. Note, that indices start with 0.
After you've created the object you can draw the similarity plot.
Call the method simgen() of the Simgen object to draw the plot. Pass the following parameters to the method:
window: sliding window size. The number of nucleotides the sliding window will span. It has the value of 500 by default.shift: this is the step our window slides downstream the alignment. It's value is set to 250 by defaultpot_rec: the index of the potential recombinant. All the other sequences will be plotted as function of distance to that sequence. Use methodget_info()to get the indices, especially if your alignment has many sequences.
The isolate of Ba genotype is the recombinant between the virus of C genotype and genotype Bj. Let's plot it. We set genotype Ba as the potential recombinant :
sim_obj.simgen(window=200, shift=50, pot_rec=1)
Potential recombinant is not shown in the plot, as the distances are calculated relative to it. The higher is the distance function (i.e. the closer to 1), the closer is the sequence to the recombinant and vice versa.
We can see typical 'crossover' of the distances which is the indicator of the possible recombination event. The distance of one isolate 'drops down' whereas the distance of the other remains the same of even gets closer to the potential recombinant, this abrupt drop shows that recombination could take place.
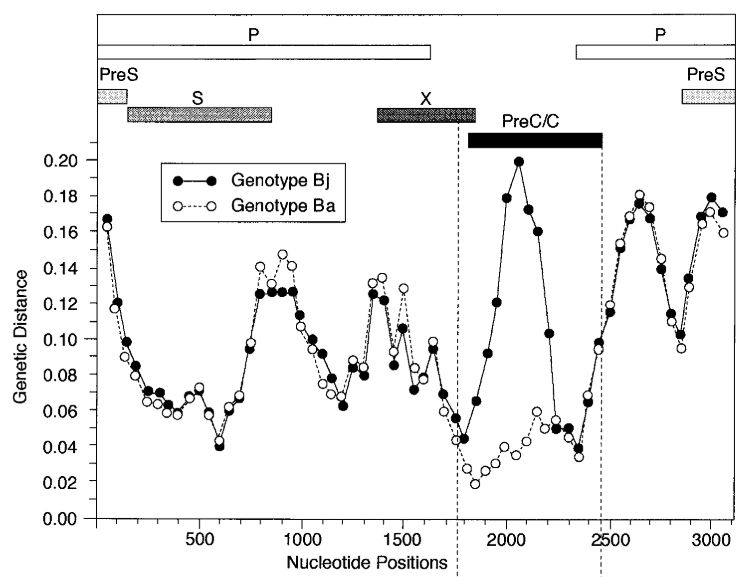
The picture from the article is shown below. It's just turned upside down relative to our plot, and instead of distance drop we see distance rising. Here Bj 'goes away' from the genotype C, whereas Ba keeps the same distance
By default simgen() method plots the whole alignment. But after initial exploration, we can take a closer look at a particular region by passing the region parameter to the simgen method. We can slice the alignment by using this parameter. region must be a tuple or a list with two integers: the start and the end position of the alignment slice.
region = (start, end)sim_obj.simgen(window=200, shift=50, pot_rec=1, region=(1000, 2700))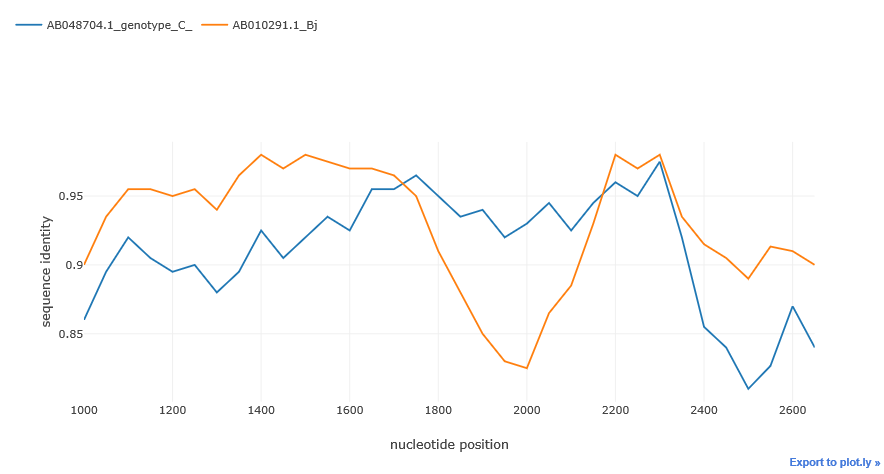
To customize the plot or just to export and store the data, use get_data() method. get_data() returns pandas DataFrame object with sequences as samples, and distances at given points as features.
sim_obj.get_data()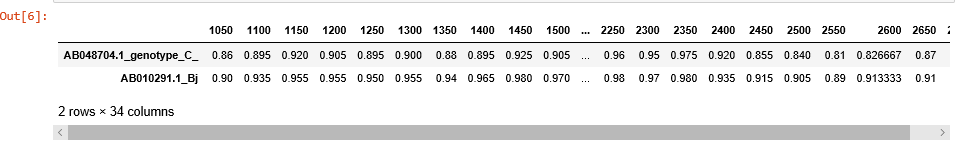
If optional paremeter df is set to False, get_data() returns a tuple containing list of ticks and a dictionary of lists. Each dictionary key is the sequence id, and lists under the keys contain the corresponding distances.
positions, data = sim_obj.get_data(df=False)print(positions)
[1050, 1100, 1150, 1200, 1250, 1300, 1350, 1400, 1450, 1500, 1550, 1600, 1650, 1700, 1750, 1800, 1850, 1900, 1950, 2000, 2050, 2100, 2150, 2200, 2250, 2300, 2350, 2400, 2450, 2500, 2550, 2600, 2650, 2700]
print(data)
{'AB048704.1_genotype_C_': [0.88, 0.935, 0.925, 0.955, 0.955, 0.965, 0.95, 0.935, 0.94, 0.92, 0.9299999999999999, 0.945, 0.925, 0.945, 0.96, 0.95, 0.975, 0.9733333333333334, 0.96, 0.96], 'AB010291.1_Bj': [0.98, 0.975, 0.97, 0.97, 0.965, 0.95, 0.91, 0.88, 0.85, 0.83, 0.825, 0.865, 0.885, 0.9299999999999999, 0.98, 0.97, 0.98, 0.9733333333333334, 0.96, 0.96]}
Once you've returned the data, you can easily customize the plot by using your favourite plotting library:
dist_data = sim_obj.get_data()
import matplotlib.pyplot as plt
import seaborn as sns
sns.set()
fig_dist1 = plt.figure(figsize=(20, 8))
plt.plot(df.loc["AB048704.1_genotype_C_", : ], lw=7, alpha=0.7, label="AB048704.1_genotype_C_")
plt.plot(df.loc["AB010291.1_Bj", : ], lw=7, alpha=0.7, label="AB010291.1_Bj")
plt.ylim(0.75, 1.05)
plt.title("similarity distance plot", fontsize=25)
plt.ylabel("distance relative to Ba", fontsize=20)
plt.xlabel("nucleotide position", fontsize=20)
plt.xticks(fontsize=15)
plt.yticks(fontsize=15)
plt.axvline(1750, alpha=0.5, color="red", lw=3,
linestyle="dashed", label="putative recombination break points")
plt.axvline(2250, alpha=0.5, color="red", lw=3,
linestyle="dashed" )
plt.legend(prop={"size":20})
plt.show()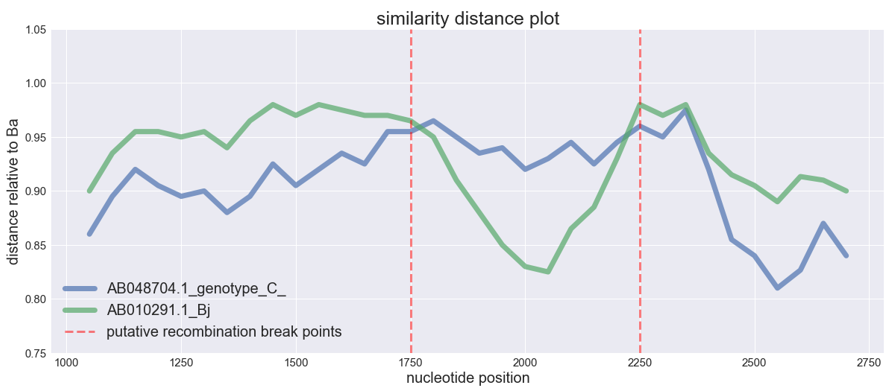
simgen() method has optional parameter dist which denoted method used to calculate pairwise distance. By default its value is set to pdist, so simgen() calculates simple pairwise distance.
Parameters for distance calculation methods:
pdist: pairwise distance (default)jcd: Jukes-Cantor distancek2p: Kimura 2-parameter distancetd: Tamura distance
sim_obj.simgen(window=200, shift=50, pot_rec=1, region=(1000, 2700), dist='k2p')to save the distance data in csv format use the method save_data():
sim_obj.save_data(out_name="hbv_distance_data")If there are about 20 or 30 sequences in the input file and their names are long, legend element may hide the plot. So, to be able to analyze many sequences at once, it's better to use short consice sequence names instead of long ones. Like this:
To illustrate how typical breakpoints may look like, here are shown some examples of previously described recombinations in the genomes of different viruses. The fasta alignments used are available at datasets folder.
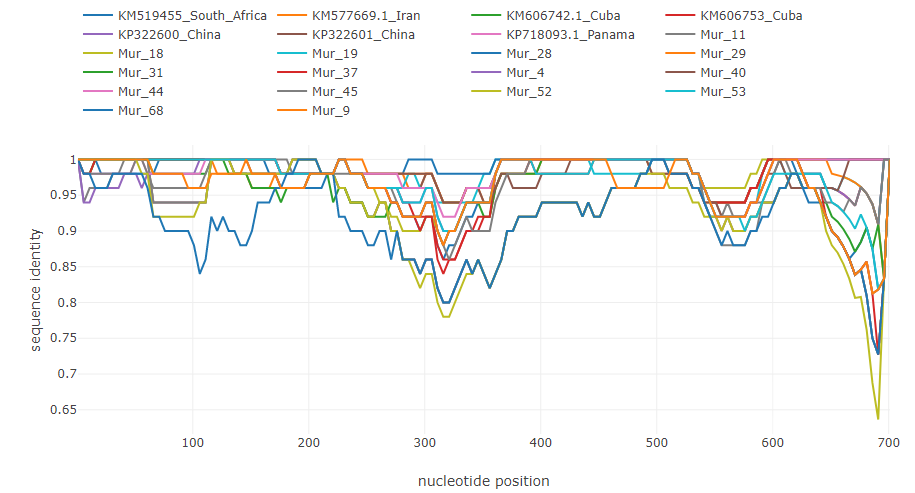
Putative recombinations in the of 145000 bp genome of lumpy skin disease virus [4]:
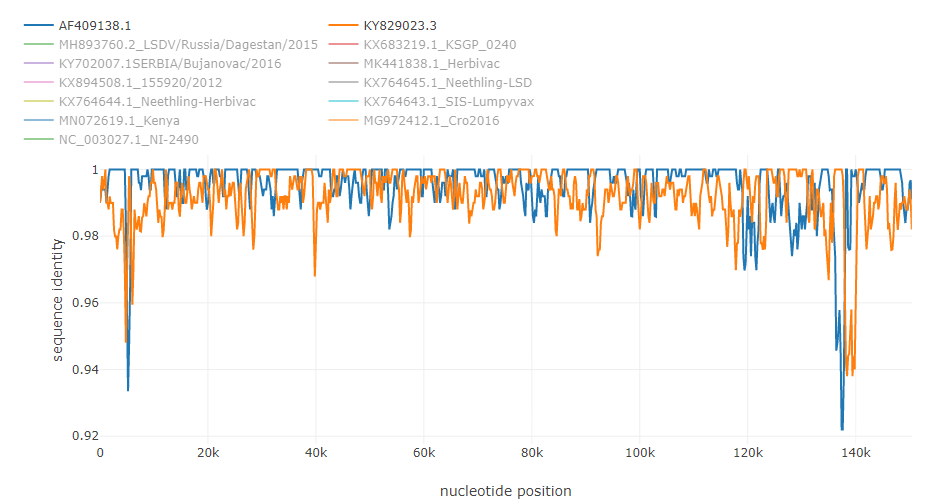
Recombination in HIV genome [5]:
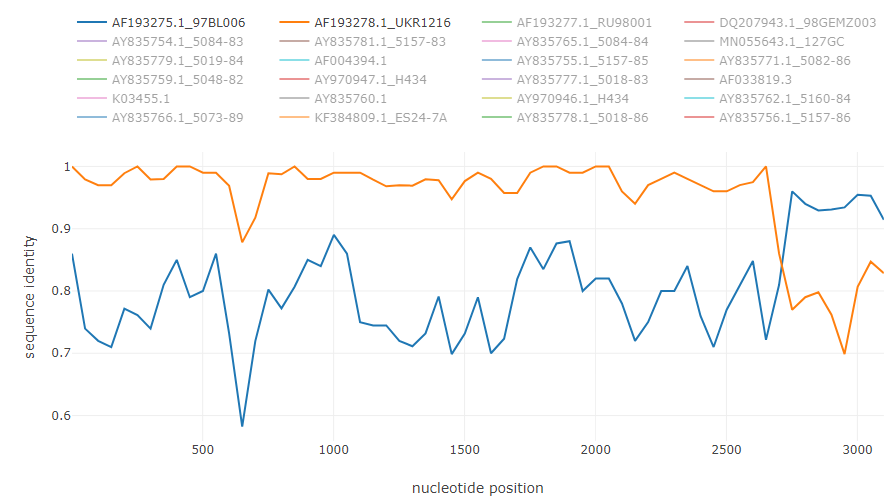
HCV intergenotype recombinant 2k/1b [6]:
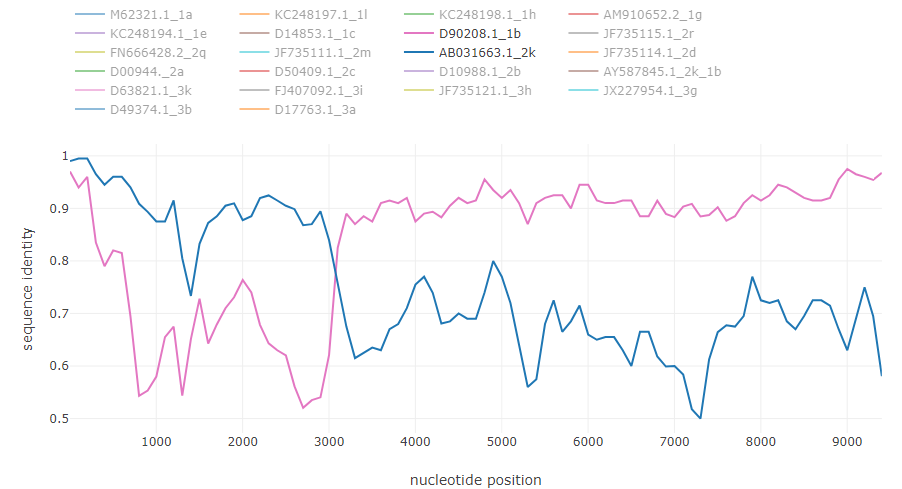
Norovirus recombinant isolate [7]:
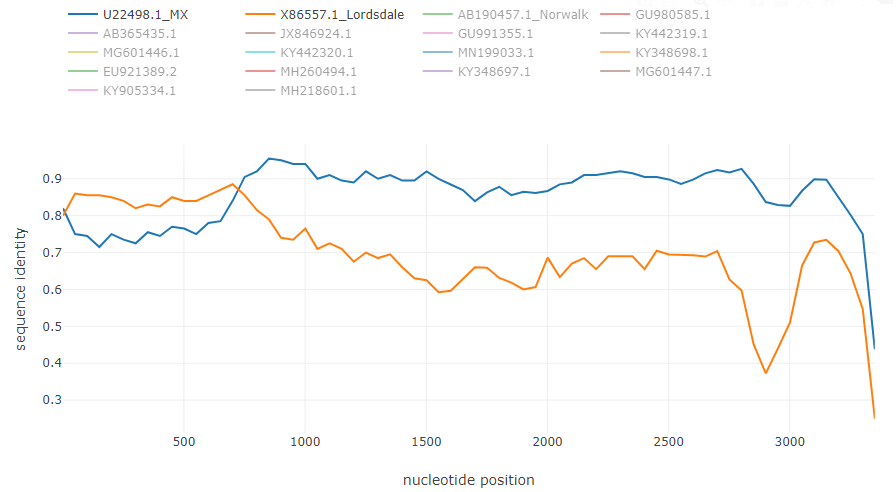
- the optimal window size is about 200-250 bp, the optimal window shift is typicall about 50-150 bp
- now distance calculation skips degenerate nucleotides and gaps and they do not influence the distance values
To verify the installation, go to the recan/test/ folder and run:
$ pytest test.py
To download the datasets use the following link: https://drive.google.com/drive/folders/1v2lg5yUDFw_fgSiulsA1uFeuzoGz0RjH?usp=sharing
- Recombination Analysis Tool (RAT): a program for the high-throughput detection of recombination. Bioinformatics, Volume 21, Issue 3, 1 February 2005, Pages 278–281, https://doi.org/10.1093/bioinformatics/bth500
- https://sray.med.som.jhmi.edu/SCRoftware/simplot/
- Hepatitis B Virus of Genotype B with or without Recombination with Genotype C over the Precore Region plus the Core Gene. Fuminaka Sugauchi et al. JOURNAL OF VIROLOGY, June 2002, p. 5985–5992. 10.1128/JVI.76.12.5985-5992.2002 https://jvi.asm.org/content/76/12/5985
- Sprygin A, Babin Y, Pestova Y, Kononova S, Wallace DB, Van Schalkwyk A, et al. (2018) Analysis and insights into recombination signals in lumpy skin disease virus recovered in the field. PLoS ONE 13(12): e0207480. https://doi.org/10.1371/journal.pone.0207480
- Liitsola, K., Holm K., Bobkov, A., Pokrovsky, V., Smolskaya,T., Leinikki,P., Osmanov,S. and Salminen,M. (2000) An AB recombinant and its parental HIV type 1 strains in the area of the former Soviet Union: low requirements for sequence identity in recombination. UNAIDS Virus Isolation Network. AIDS Res. Hum. Retroviruses, 16, 1047–1053.
- Smith, D. B., Bukh, J., Kuiken, C., Muerhoff, A. S., Rice, C. M., Stapleton, J. T., & Simmonds, P. (2014). Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology, 59(1), 318–327. https://doi.org/10.1002/hep.26744
- Jiang,X., Espul,C., Zhong,W.M., Cuello,H. and Matson,D.O. (1999) Characterization of a novel human calicivirus that may be a naturally occurring recombinant. Arch. Virol., 144, 2377–2387.
- Martin, D. P., Murrell, B., Golden, M., Khoosal, A., & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1(1), 1–5. https://doi.org/10.1093/ve/vev003
- Babin, Y., (2020). Recan: Python tool for analysis of recombination events in viral genomes. Journal of Open Source Software, 5(49), 2014. https://doi.org/10.21105/joss.02014
- Characterization of SARS-CoV-2 P.1 (Gamma) Variant of Concern From Amazonas, Brazil. Zimerman RA et al. (2022). Comparative Genomics and Front. Med. 9:806611. https://doi.org/10.3389/fmed.2022.806611
- In book: Proceedings of the 4th International Conference on Big Data Analytics for Cyber-Physical System in Smart City - Volume 2. Chapter: Python Data Analysis Techniques in Administrative Information Integration Management System April 2023 https://doi.org/10.1007/978-981-99-1157-8_35
- Substantial viral diversity in bats and rodents from East Africa: insights into evolution, recombination, and cocirculation. Daxi Wang et al. 2024. Microbiome (2024) 12:72 https://doi.org/10.1186/s40168-024-01782-4
- Identification of Recombinant Aichivirus D in Cattle, Italy.Pellegrini, F et al. I Animals 2024, 14, 3315. https://doi.org/10.3390/ani14223315