

Mechanisms that regulate Epstein-Barr virus EBNA-1 gene transcription during restricted latency are conserved among lymphocryptoviruses of Old World primates
- PMID: 9971778
- PMCID: PMC104440
- DOI: 10.1128/JVI.73.3.1980-1989.1999
Mechanisms that regulate Epstein-Barr virus EBNA-1 gene transcription during restricted latency are conserved among lymphocryptoviruses of Old World primates
Abstract
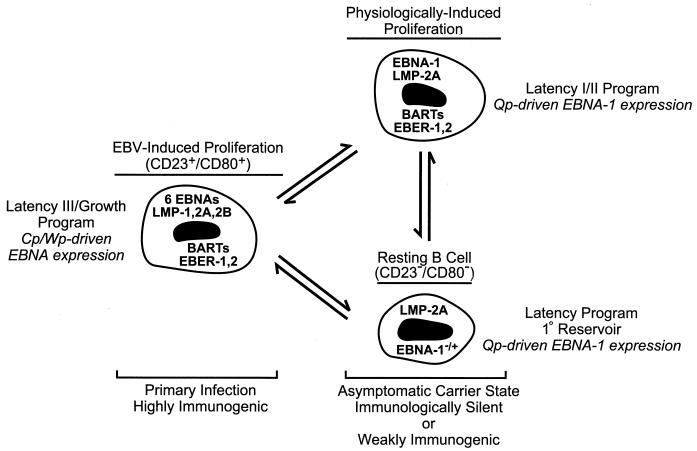
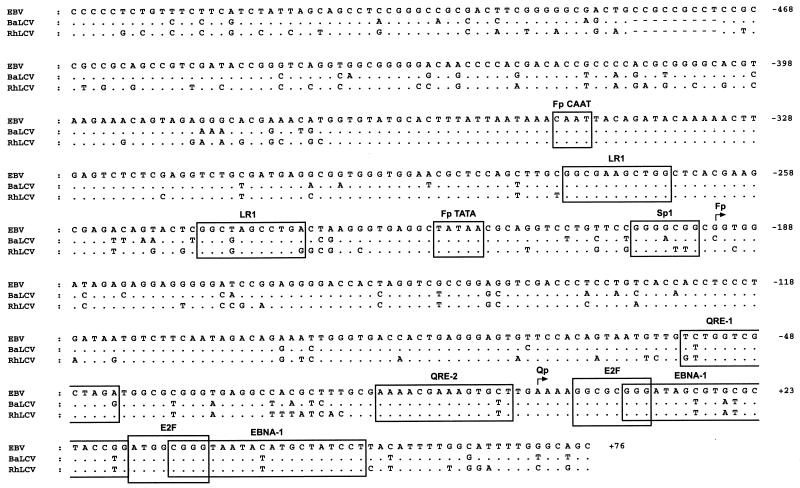
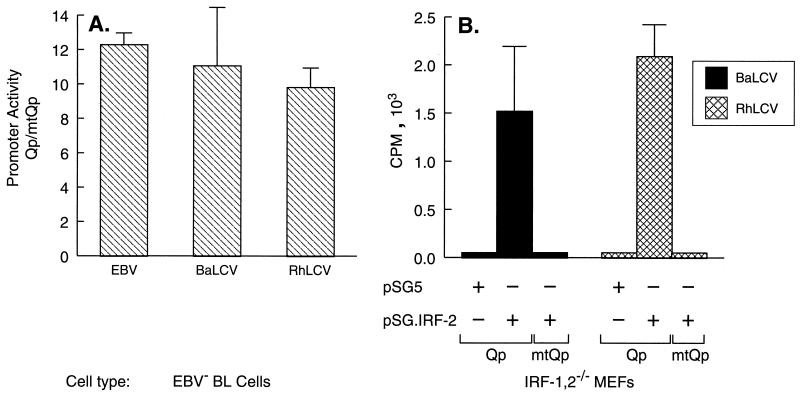
Epstein-Barr virus (EBV), the only known human lymphocryptovirus (LCV), displays a remarkable degree of genetic and biologic identity to LCVs that infect Old World primates. Within their natural hosts, infection by these viruses recapitulates many key aspects of EBV infection, including the establishment of long-term latency within B lymphocytes, and is therefore a potentially valuable animal model of EBV infection. However, it is unclear whether these LCVs have adopted or maintained the same mechanisms used by EBV to express essential viral proteins, such as EBNA-1, in the face of cell-mediated repression of EBV gene expression that occurs upon establishment of the asymptomatic carrier state. To address this issue, we determined whether the endogenous LCVs of baboon (Cercopithecine herpesvirus 12) and rhesus macaque (Cercopithecine herpesvirus 15) have the functional equivalent of the EBV promoter Qp, which mediates exclusive expression of EBNA-1 during the restricted programs of EBV latency associated with the carrier state. Our results indicate that (i) both the baboon and rhesus macaque LCVs have a genomic locus that is highly homologous to the EBV Qp region, (ii) key cis-regulatory elements of Qp are conserved in these LCV genomes and compose promoters that are functionally indistinguishable from EBV Qp, and (iii) EBNA-1 transcripts identical in structure to EBV Qp-specific EBNA-1 mRNAs are present in nonhuman LCV-infected cells, demonstrating that these Qp homologs are indeed utilized as alternative EBNA-1 promoters. These observations indicate that the molecular mechanisms which regulate EBV gene expression during restricted latency have been conserved among the LCVs. The contribution of these mechanisms to viral persistence in vivo can now be experimentally tested in nonhuman primate models of LCV infection.
Figures




Similar articles
-
Transcriptional regulatory properties of Epstein-Barr virus nuclear antigen 3C are conserved in simian lymphocryptoviruses.J Virol. 2003 May;77(10):5639-48. doi: 10.1128/jvi.77.10.5639-5648.2003. J Virol. 2003. PMID: 12719556 Free PMC article.
-
Sequence and functional analysis of EBNA-LP and EBNA2 proteins from nonhuman primate lymphocryptoviruses.J Virol. 2000 Jan;74(1):379-89. doi: 10.1128/jvi.74.1.379-389.2000. J Virol. 2000. PMID: 10590127 Free PMC article.
-
Infection of human B lymphocytes with lymphocryptoviruses related to Epstein-Barr virus.J Virol. 1998 Apr;72(4):3205-12. doi: 10.1128/JVI.72.4.3205-3212.1998. J Virol. 1998. PMID: 9525646 Free PMC article.
-
Non-human Primate Lymphocryptoviruses: Past, Present, and Future.Curr Top Microbiol Immunol. 2015;391:385-405. doi: 10.1007/978-3-319-22834-1_13. Curr Top Microbiol Immunol. 2015. PMID: 26428382 Review.
-
Simian homologues of Epstein-Barr virus.Philos Trans R Soc Lond B Biol Sci. 2001 Apr 29;356(1408):489-97. doi: 10.1098/rstb.2000.0776. Philos Trans R Soc Lond B Biol Sci. 2001. PMID: 11313007 Free PMC article. Review.
Cited by
-
Epigenetic regulation of EBV and KSHV latency.Curr Opin Virol. 2013 Jun;3(3):251-9. doi: 10.1016/j.coviro.2013.03.004. Epub 2013 Apr 16. Curr Opin Virol. 2013. PMID: 23601957 Free PMC article. Review.
-
Strong selective pressure for evolution of an Epstein-Barr virus LMP2B homologue in the rhesus lymphocryptovirus.J Virol. 1999 Oct;73(10):8867-72. doi: 10.1128/JVI.73.10.8867-8872.1999. J Virol. 1999. PMID: 10482645 Free PMC article.
-
LANA oligomeric architecture is essential for KSHV nuclear body formation and viral genome maintenance during latency.PLoS Pathog. 2019 Jan 25;15(1):e1007489. doi: 10.1371/journal.ppat.1007489. eCollection 2019 Jan. PLoS Pathog. 2019. PMID: 30682185 Free PMC article.
-
Inhibition of antigen presentation by the glycine/alanine repeat domain is not conserved in simian homologues of Epstein-Barr virus nuclear antigen 1.J Virol. 1999 Sep;73(9):7381-9. doi: 10.1128/JVI.73.9.7381-7389.1999. J Virol. 1999. PMID: 10438828 Free PMC article.
-
Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1.J Biomol Screen. 2010 Oct;15(9):1107-15. doi: 10.1177/1087057110379154. J Biomol Screen. 2010. PMID: 20930215 Free PMC article.
References
-
- Ablashi D V, Gerber P, Easton J. Oncogenic herpesviruses of nonhuman primates. Comp Immunol Microbiol Infect Dis. 1979;2:229–241. - PubMed
-
- Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: John Wiley and Sons, Inc.; 1989.
-
- Baer R, Bankier A T, Biggin M D, Deininger P L, Farrell P J, Gibson T G, Hatfull G, Hudson G S, Satchwell S C, Sequin C, Tufnell P S, Barrell B G. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310:207–211. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
