

The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small
- PMID: 9770497
- PMCID: PMC22842
- DOI: 10.1073/pnas.95.21.12390
The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small
Abstract
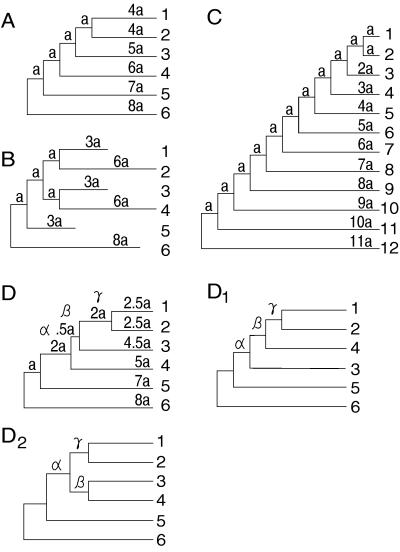
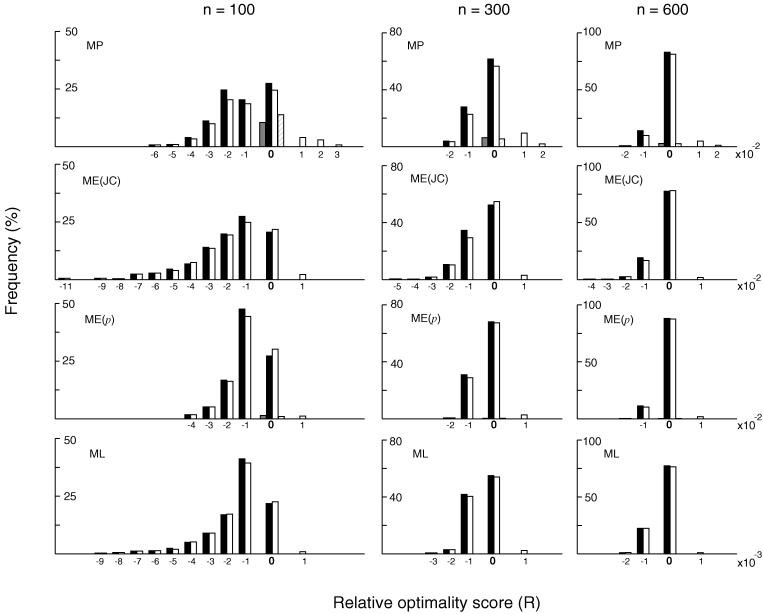
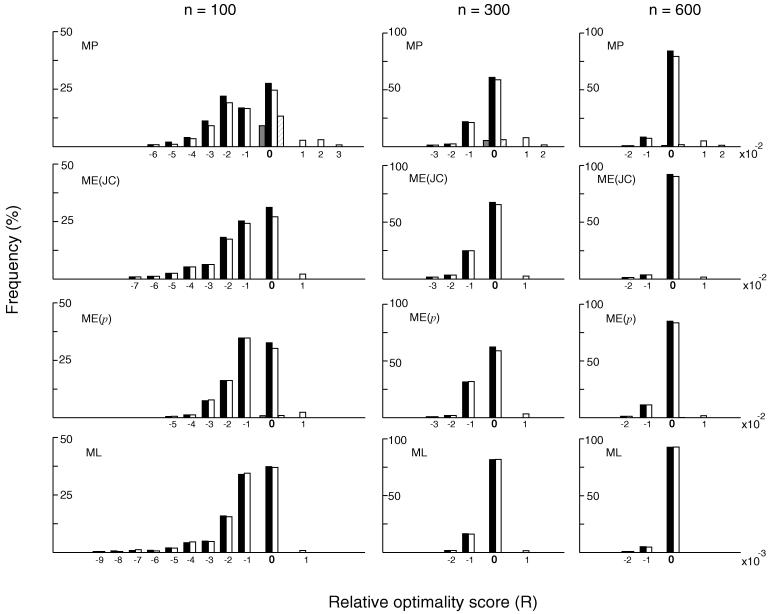
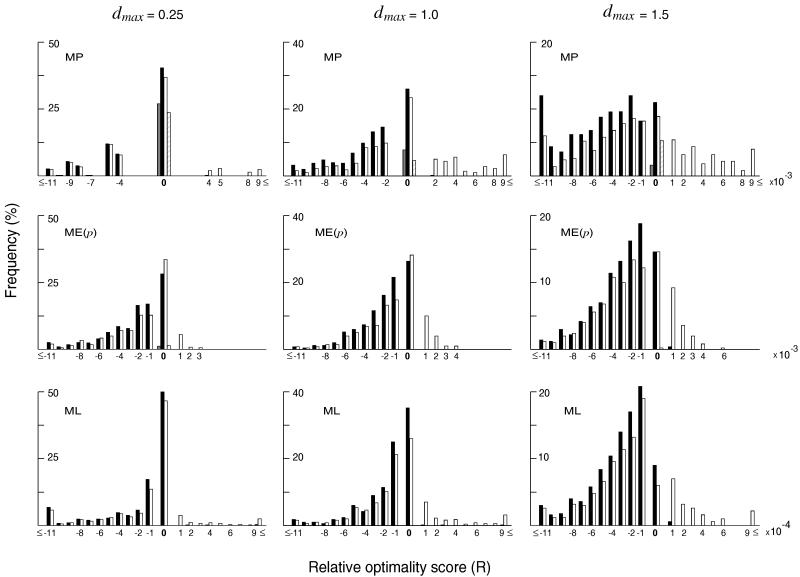
In the maximum parsimony (MP) and minimum evolution (ME) methods of phylogenetic inference, evolutionary trees are constructed by searching for the topology that shows the minimum number of mutational changes required (M) and the smallest sum of branch lengths (S), respectively, whereas in the maximum likelihood (ML) method the topology showing the highest maximum likelihood (A) of observing a given data set is chosen. However, the theoretical basis of the optimization principle remains unclear. We therefore examined the relationships of M, S, and A for the MP, ME, and ML trees with those for the true tree by using computer simulation. The results show that M and S are generally greater for the true tree than for the MP and ME trees when the number of nucleotides examined (n) is relatively small, whereas A is generally lower for the true tree than for the ML tree. This finding indicates that the optimization principle tends to give incorrect topologies when n is small. To deal with this disturbing property of the optimization principle, we suggest that more attention should be given to testing the statistical reliability of an estimated tree rather than to finding the optimal tree with excessive efforts. When a reliability test is conducted, simplified MP, ME, and ML algorithms such as the neighbor-joining method generally give conclusions about phylogenetic inference very similar to those obtained by the more extensive tree search algorithms.
Figures
Similar articles
-
Efficiencies of fast algorithms of phylogenetic inference under the criteria of maximum parsimony, minimum evolution, and maximum likelihood when a large number of sequences are used.Mol Biol Evol. 2000 Aug;17(8):1251-8. doi: 10.1093/oxfordjournals.molbev.a026408. Mol Biol Evol. 2000. PMID: 10908645
-
A rapid heuristic algorithm for finding minimum evolution trees.Mol Phylogenet Evol. 2000 Aug;16(2):173-9. doi: 10.1006/mpev.1999.0728. Mol Phylogenet Evol. 2000. PMID: 10942605
-
Efficiencies of different genes and different tree-building methods in recovering a known vertebrate phylogeny.Mol Biol Evol. 1996 Mar;13(3):525-36. doi: 10.1093/oxfordjournals.molbev.a025613. Mol Biol Evol. 1996. PMID: 8742641
-
Phylogenetic analysis in molecular evolutionary genetics.Annu Rev Genet. 1996;30:371-403. doi: 10.1146/annurev.genet.30.1.371. Annu Rev Genet. 1996. PMID: 8982459 Review.
-
Distance-based phylogenetic inference from typing data: a unifying view.Brief Bioinform. 2021 May 20;22(3):bbaa147. doi: 10.1093/bib/bbaa147. Brief Bioinform. 2021. PMID: 32734294 Review.
Cited by
-
Incomplete taxon sampling is not a problem for phylogenetic inference.Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10751-6. doi: 10.1073/pnas.191248498. Epub 2001 Aug 28. Proc Natl Acad Sci U S A. 2001. PMID: 11526218 Free PMC article.
-
Fast NJ-like algorithms to deal with incomplete distance matrices.BMC Bioinformatics. 2008 Mar 26;9:166. doi: 10.1186/1471-2105-9-166. BMC Bioinformatics. 2008. PMID: 18366787 Free PMC article.
-
Calculibacillus koreensis gen. nov., sp. nov., an anaerobic Fe(III)-reducing bacterium isolated from sediment of mine tailings.J Microbiol. 2016 Jun;54(6):413-9. doi: 10.1007/s12275-016-6086-8. Epub 2016 May 27. J Microbiol. 2016. PMID: 27225457
-
Mucilaginibacter aquariorum sp. nov., Isolated from Fresh Water.J Microbiol Biotechnol. 2022 Dec 28;32(12):1553-1560. doi: 10.4014/jmb.2208.08021. Epub 2022 Oct 31. J Microbiol Biotechnol. 2022. PMID: 36377201 Free PMC article.
-
Genetic variability and evolutionary diversification of membrane ABC transporters in plants.BMC Plant Biol. 2015 Feb 13;15:51. doi: 10.1186/s12870-014-0323-2. BMC Plant Biol. 2015. PMID: 25850033 Free PMC article.
References
-
- Eck R V, Dayhoff M O. Atlas of Protein Sequence and Structure. Silver Spring, MD: National Biomedical Research Foundation; 1966.
-
- Fitch W M. Syst Zool. 1971;20:406–416.
-
- Sober E. Reconstructing the Past: Parsimony, Evolution, and Inference. Cambridge, MA: MIT Press; 1988.
-
- Edwards A W F, Cavalli-Sforza L L. Heredity. 1963;18:553. (abstr.).
-
- Saitou N, Imanishi M. Mol Biol Evol. 1989;6:514–525.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
