

Functional interference of Sp1 and NF-kappaB through the same DNA binding site
- PMID: 9488441
- PMCID: PMC108839
- DOI: 10.1128/MCB.18.3.1266
Functional interference of Sp1 and NF-kappaB through the same DNA binding site
Abstract
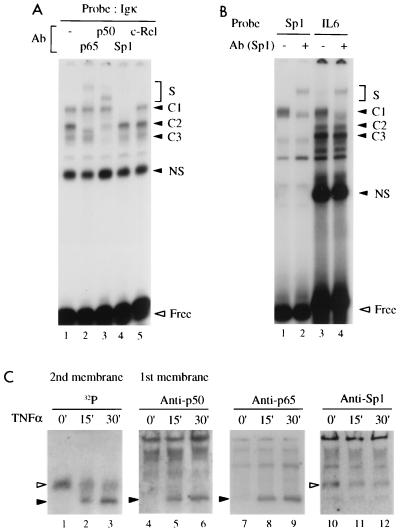
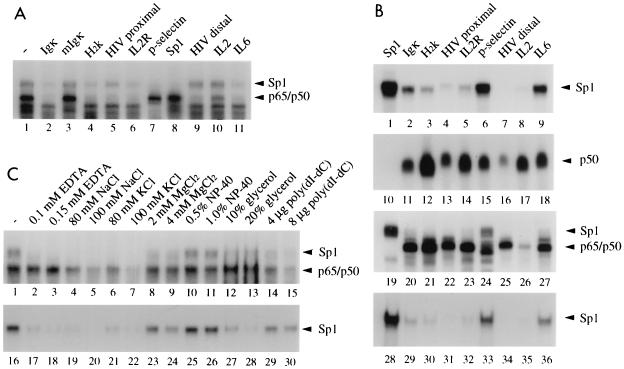
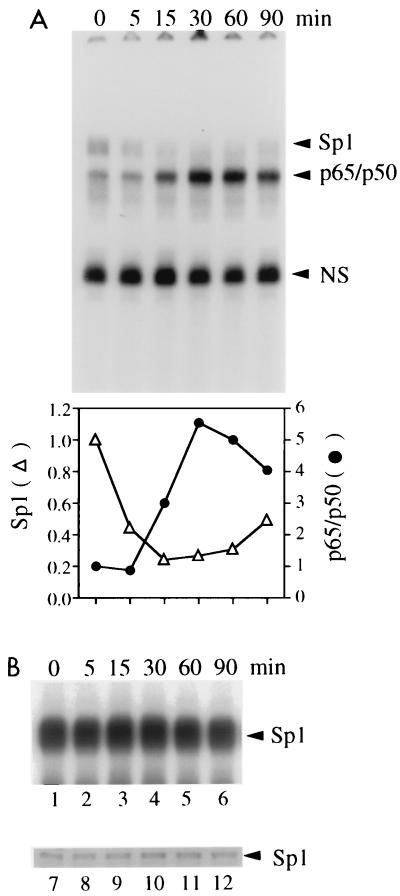
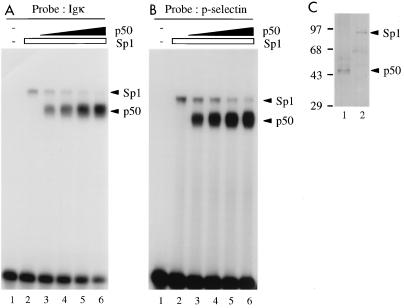
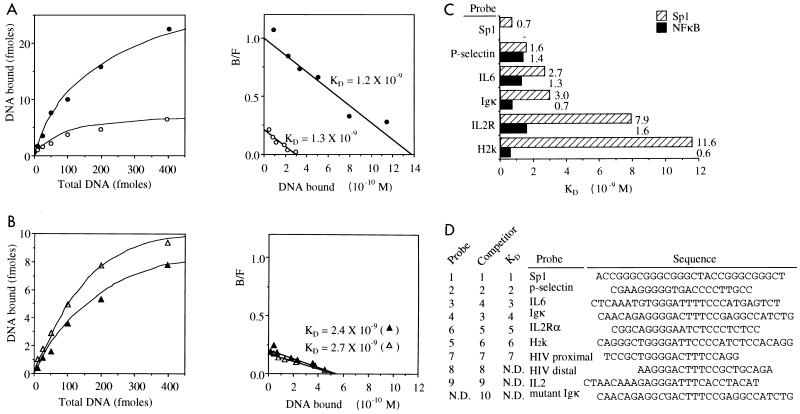
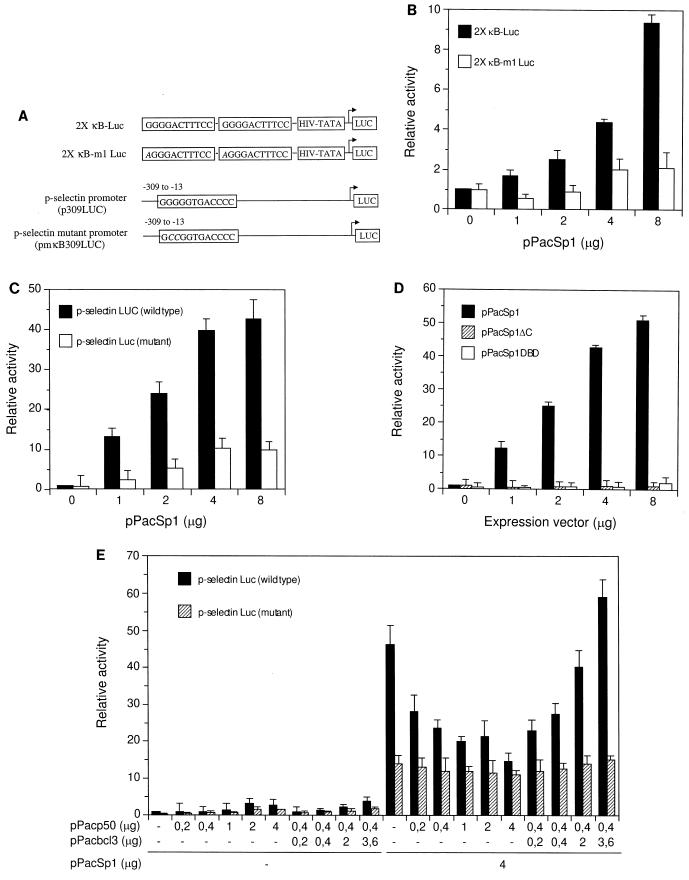
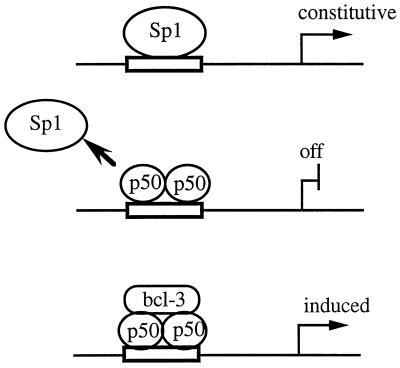
Gene activation by NF-kappaB/Rel transcription factors is modulated by synergistic or antagonistic interactions with other promoter-bound transcription factors. For example, Sp1 sites are often found in NF-kappaB-regulated genes, and Sp1 can activate certain promoters in synergism with NF-kappaB through nonoverlapping binding sites. Here we report that Sp1 acts directly through a subset of NF-kappaB binding sites. The DNA binding affinity of Sp1 to these NF-kappaB sites, as determined by their relative dissociation constants and their relative efficiencies as competitor DNAs or as binding site probes, is in the order of that for a consensus GC box Sp1 site. In contrast, NF-kappaB does not bind to a GC box Sp1 site. Sp1 can activate transcription through immunoglobulin kappa-chain enhancer or P-selectin promoter NF-kappaB sites. p50 homodimers replace Sp1 from the P-selectin promoter by binding site competition and thereby either inhibit basal Sp1-driven expression or, in concert with Bcl-3, stimulate expression. The interaction of Sp1 with NF-kappaB sites thus provides a means to keep an elevated basal expression of NF-kappaB-dependent genes in the absence of activated nuclear NF-kappaB/Rel.
Figures
Similar articles
-
Differential regulation of CCL22 gene expression in murine dendritic cells and B cells.J Immunol. 2005 May 1;174(9):5620-9. doi: 10.4049/jimmunol.174.9.5620. J Immunol. 2005. PMID: 15843561
-
Transcriptional activation of TINF2, a gene encoding the telomere-associated protein TIN2, by Sp1 and NF-κB factors.PLoS One. 2011;6(6):e21333. doi: 10.1371/journal.pone.0021333. Epub 2011 Jun 23. PLoS One. 2011. PMID: 21731707 Free PMC article.
-
Characterization of the human intestinal CD98 promoter and its regulation by interferon-gamma.Am J Physiol Gastrointest Liver Physiol. 2007 Feb;292(2):G535-45. doi: 10.1152/ajpgi.00385.2006. Epub 2006 Oct 5. Am J Physiol Gastrointest Liver Physiol. 2007. PMID: 17023546
-
Unique aspects of transcriptional regulation in neurons--nuances in NFkappaB and Sp1-related factors.J Neuroinflammation. 2009 May 18;6:16. doi: 10.1186/1742-2094-6-16. J Neuroinflammation. 2009. PMID: 19450264 Free PMC article. Review.
-
Genome reading by the NF-κB transcription factors.Nucleic Acids Res. 2019 Nov 4;47(19):9967-9989. doi: 10.1093/nar/gkz739. Nucleic Acids Res. 2019. PMID: 31501881 Free PMC article. Review.
Cited by
-
Nuclear factor κB mediates suppression of canonical transient receptor potential 6 expression by reactive oxygen species and protein kinase C in kidney cells.J Biol Chem. 2013 May 3;288(18):12852-65. doi: 10.1074/jbc.M112.410357. Epub 2013 Mar 22. J Biol Chem. 2013. PMID: 23525112 Free PMC article.
-
Chronic exposure to 12-O-tetradecanoylphorbol-13-acetate represses sod2 induction in vivo: the negative role of p50.Carcinogenesis. 2007 Dec;28(12):2605-13. doi: 10.1093/carcin/bgm163. Epub 2007 Jul 25. Carcinogenesis. 2007. PMID: 17652337 Free PMC article.
-
Crystal structure of the ankyrin repeat domain of Bcl-3: a unique member of the IkappaB protein family.EMBO J. 2001 Nov 15;20(22):6180-90. doi: 10.1093/emboj/20.22.6180. EMBO J. 2001. PMID: 11707390 Free PMC article.
-
The role of Sp1 in IL-1beta and H. pylori-mediated regulation of H,K-ATPase gene transcription.Am J Physiol Gastrointest Liver Physiol. 2008 Nov;295(5):G977-86. doi: 10.1152/ajpgi.90338.2008. Epub 2008 Sep 4. Am J Physiol Gastrointest Liver Physiol. 2008. PMID: 18772363 Free PMC article.
-
Alteration in the activation state of new inflammation-associated targets by phospholipase A2-activating protein (PLAA).Cell Signal. 2008 May;20(5):844-61. doi: 10.1016/j.cellsig.2008.01.004. Epub 2008 Jan 17. Cell Signal. 2008. PMID: 18291623 Free PMC article.
References
-
- Augustin L B, Felsheim R F, Min B H, Fuchs S M, Fuchs J A, Loh H H. Genomic structure of the mouse delta opioid receptor gene. Biochem Biophys Res Commun. 1995;207:111–119. - PubMed
-
- Baeuerle P A, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. - PubMed
-
- Ballard D W, Bohnlein E, Lowenthal J W, Wano Y, Franza B R, Greene W C. HTLV-I tax induces cellular proteins that activate the κB element in the IL-2 receptor α gene. Science. 1988;241:1652–1655. - PubMed
-
- Beg A A, Baldwin A S., Jr The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev. 1993;7:2064–2070. - PubMed
-
- Beg A A, Sha W C, Bronson R T, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous