

Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx
- PMID: 9482940
- PMCID: PMC19446
- DOI: 10.1073/pnas.95.5.2642
Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx
Abstract
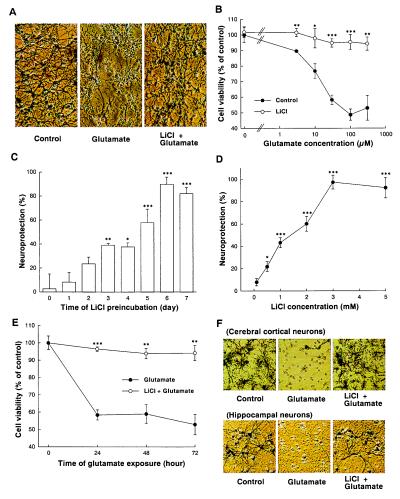
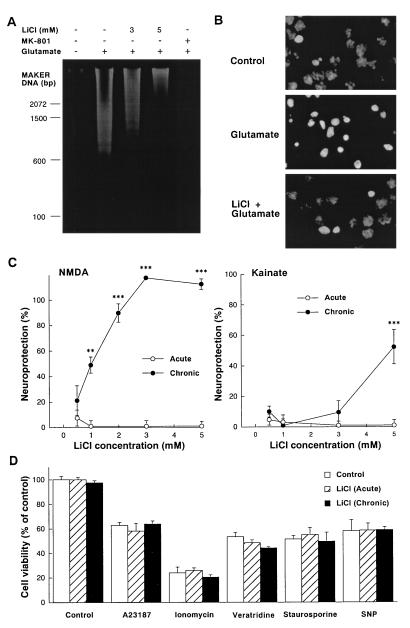
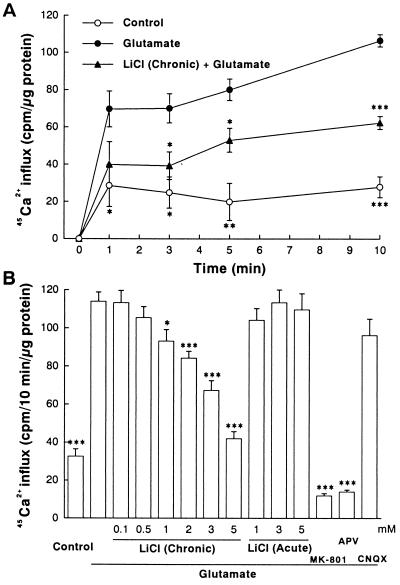
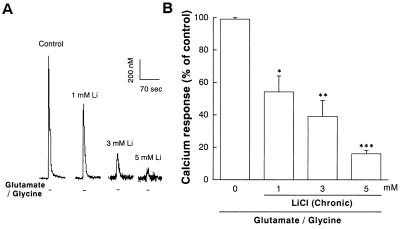
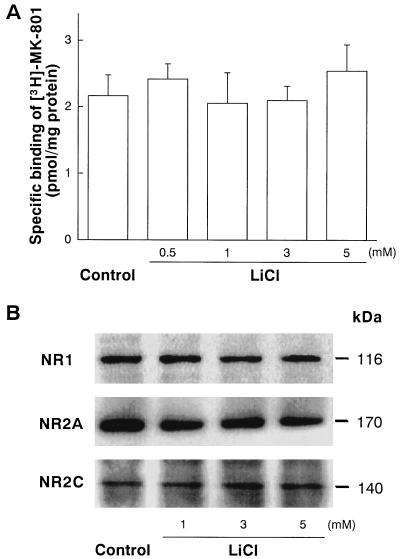
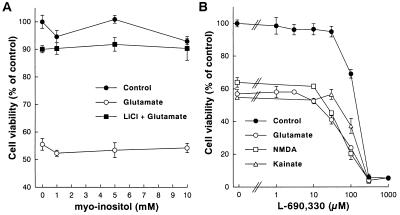
Lithium is the most commonly used drug for the treatment of manic depressive illness. The precise mechanisms underlying its clinical efficacy remain unknown. We found that long-term exposure to lithium chloride dramatically protects cultured rat cerebellar, cerebral cortical, and hippocampal neurons against glutamate-induced excitotoxicity, which involves apoptosis mediated by N-methyl-D-aspartate (NMDA) receptors. This neuroprotection is long-lasting, occurs at therapeutically relevant concentrations of lithium with an EC50 of approximately 1.3 mM, and requires treatment for 6-7 days for complete protection to occur. In contrast, a 24-h treatment with lithium is ineffective. The protection in cerebellar neurons is specific for glutamate-induced excitotoxicity and can be attributed to inhibition of NMDA receptor-mediated calcium influx measured by 45Ca2+ uptake studies and fura-2 fluorescence microphotometry. The long-term effects of lithium are not caused by down-regulation of NMDA receptor subunit proteins and are unlikely related to its known ability to block inositol monophosphatase activity. Our results suggest that modulation of glutamate receptor hyperactivity represents at least part of the molecular mechanisms by which lithium alters brain function and exerts its clinical efficacy in the treatment for manic depressive illness. These actions of lithium also suggest that abnormality of glutamatergic neurotransmission as a pathogenic mechanism underlying bipolar illness warrants future investigation.
Figures
Similar articles
-
The excitoprotective effect of N-methyl-D-aspartate receptors is mediated by a brain-derived neurotrophic factor autocrine loop in cultured hippocampal neurons.J Neurochem. 2005 Aug;94(3):713-22. doi: 10.1111/j.1471-4159.2005.03200.x. Epub 2005 Jul 5. J Neurochem. 2005. PMID: 16000165
-
N-methyl-D-aspartate receptor-mediated mitochondrial Ca(2+) overload in acute excitotoxic motor neuron death: a mechanism distinct from chronic neurotoxicity after Ca(2+) influx.J Neurosci Res. 2001 Mar 1;63(5):377-87. doi: 10.1002/1097-4547(20010301)63:5<377::AID-JNR1032>3.0.CO;2-#. J Neurosci Res. 2001. PMID: 11223912
-
Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation.J Neurochem. 2002 Feb;80(4):589-97. doi: 10.1046/j.0022-3042.2001.00728.x. J Neurochem. 2002. PMID: 11841566
-
[Neuroprotective actions of lithium].Seishin Shinkeigaku Zasshi. 2003;105(1):81-6. Seishin Shinkeigaku Zasshi. 2003. PMID: 12701214 Review. Japanese.
-
Neuroprotective effects of lithium in cultured cells and animal models of diseases.Bipolar Disord. 2002 Apr;4(2):129-36. doi: 10.1034/j.1399-5618.2002.01179.x. Bipolar Disord. 2002. PMID: 12071510 Review.
Cited by
-
Deleterious effect of LiCl on honeybee (Aphis mellifera) grubs and no effect on Varroa mites (Varroa destructor) under normal beekeeping management.Biol Futur. 2024 Jun;75(2):199-204. doi: 10.1007/s42977-023-00196-x. Epub 2023 Dec 6. Biol Futur. 2024. PMID: 38055159
-
Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics.Mol Neurobiol. 2005 Oct;32(2):173-202. doi: 10.1385/MN:32:2:173. Mol Neurobiol. 2005. PMID: 16215281 Review.
-
Potential mechanisms underlying lithium treatment for Alzheimer's disease and COVID-19.Eur Rev Med Pharmacol Sci. 2022 Mar;26(6):2201-2214. doi: 10.26355/eurrev_202203_28369. Eur Rev Med Pharmacol Sci. 2022. PMID: 35363371 Free PMC article. Review.
-
Round-window delivery of lithium chloride regenerates cochlear synapses damaged by noise-induced excitotoxic trauma via inhibition of the NMDA receptor in the rat.PLoS One. 2023 May 22;18(5):e0284626. doi: 10.1371/journal.pone.0284626. eCollection 2023. PLoS One. 2023. PMID: 37216352 Free PMC article.
-
Targeting Tau to Treat Clinical Features of Huntington's Disease.Front Neurol. 2020 Nov 19;11:580732. doi: 10.3389/fneur.2020.580732. eCollection 2020. Front Neurol. 2020. PMID: 33329322 Free PMC article. Review.
References
-
- Goodwin F K, Jamison K R. Manic-Depressive Illness. New York: Oxford Univ. Press; 1990.
-
- Birch N J. Lithium and the Cell: Pharmacology and Biochemistry. San Diego: Academic; 1991.
-
- Post R M, Weiss S R B, Chuang D-M. J Clin Psychopharmacol. 1992;12:23S–35S. - PubMed
-
- Collingridge G L, Watkins J C. The NMDA Receptor. New York: Oxford Univ. Press; 1994.
-
- Collingridge G L, Bliss T V. Trends Neurosci. 1995;18:54–56. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
