

doi: 10.1038/s41467-024-53759-4.
A long-context language model for deciphering and generating bacteriophage genomes
Affiliations
- PMID: 39477977
- PMCID: PMC11525655
- DOI: 10.1038/s41467-024-53759-4
Item in Clipboard
A long-context language model for deciphering and generating bacteriophage genomes
Nat Commun.
.
Abstract
Inspired by the success of large language models (LLMs), we develop a long-context generative model for genomes. Our multiscale transformer model, megaDNA, is pre-trained on unannotated bacteriophage genomes with nucleotide-level tokenization. We demonstrate the foundational capabilities of our model including the prediction of essential genes, genetic variant effects, regulatory element activity and taxonomy of unannotated sequences. Furthermore, it generates de novo sequences up to 96 K base pairs, which contain potential regulatory elements and annotated proteins with phage-related functions.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
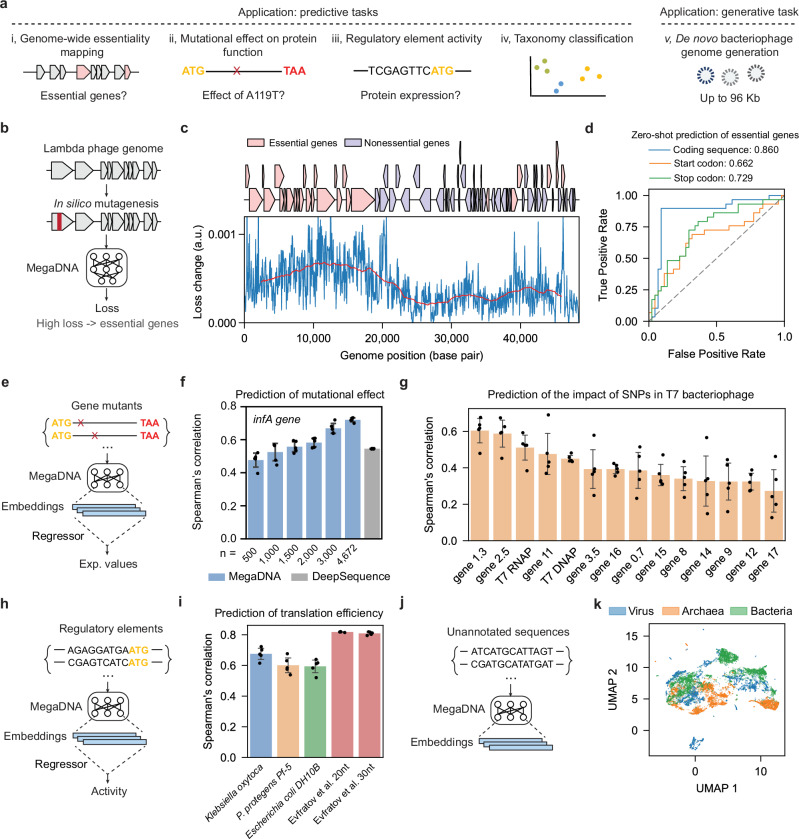
a Overview of the model applications. b In silico mutagenesis analysis to identify essential genes in the bacteriophage genome. c Model loss variation across the lambda phage genome in the mutagenesis analysis. Upper, essential, and non-essential genes in the genome. Lower: changes in model loss for 50 bp non-overlapping windows across the genome (blue). The step size is 50 bp, and moving averages of model loss across 5000 bp windows are denoted in red. d Zero-shot prediction of essential genes by calculating the effects of mutations in the gene coding region (blue), start codon (orange) and stop codon (green). Area under the ROC curve (AUROC) scores are reported. e Prediction of mutational effects on protein functions using model embeddings. f Prediction of mutational effects for the deep mutational scanning experiment of the infA gene. Spearman correlation coefficients of the predicted and reported fitness from fivefold cross-validation tests are reported (Blue: megaDNA, gray: DeepSequence). n is the number of training samples. g Prediction of the impacts of Single Nucleotide Polymorphisms (SNPs) in the T7 bacteriophage genome. Spearman correlation of the predicted and reported fitness from fivefold cross-validation tests is reported. h Prediction of regulatory element activity using model embeddings. i Prediction of translation efficiencies for non-model organisms and high-throughput gene expression libraries. For K. oxytoca, P. protegens, and E. coli DH10B, we evaluated the model performance on endogenous genes. Fivefold cross-validation tests were used for all calculations. j Classifying taxonomies of unannotated sequences using model embeddings. k UMAP visualization of model embeddings for sequences from bacteriophages, bacteria, and archaea (model middle layer, sample size: n = 5000 per group). For f, g, and i, data are presented as mean values ± SEM from fivefold cross-validation tests (n = 5 folds). Source data are provided as a Source Data file.
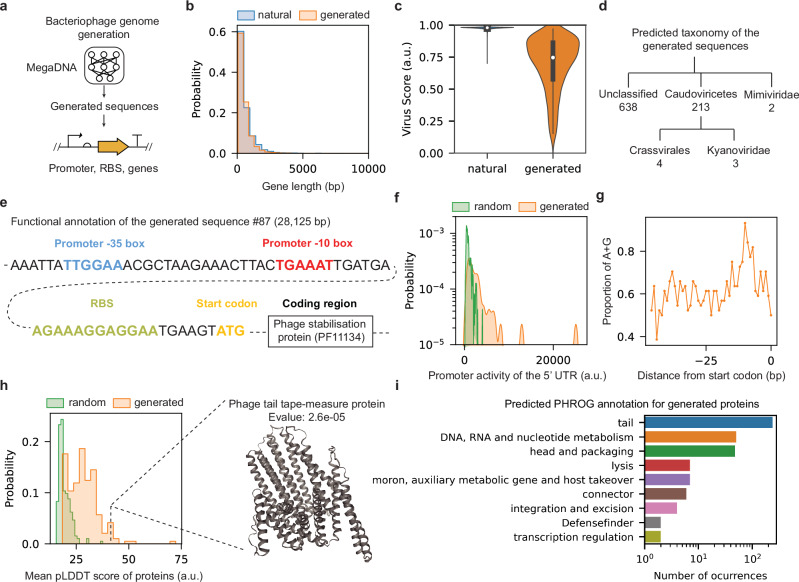
a The workflow schematic. b Comparison of gene length distributions between randomly sampled subsets of predicted genes in generated sequences and training dataset (sample size: n = 2000). Two-sided Kolmogorov–Smirnov test: p value = 0.15. c Comparison of the predicted virus scores for generated sequences (sample size: n = 1024) and the training dataset (sample size: n = 99,429). Median virus scores are indicated by white dots. Black bars denote the interquartile ranges (25th to 75th percentiles). d Predicted taxonomy for the generated sequences predicted as viral. Only taxonomies with >1 sequence are shown. geNomad was used to produce results in (c, d). e Functional annotation of a selected sequence fragment (generated sequence #87). f Predicted promoter activity for all the 5′UTRs in the generated sequence #87 (sample size: n = 44), along with the promoter activity of the random sequences with the same length. Promoter activities were calculated using the Promoter Calculator. Two-sided Kolmogorov–Smirnov test: p value = 6.3 × 10−8. g Proportions of adenine (A) and guanine (G) nucleotides preceding the start codon of all the predicted genes in the generated sequence #87. h Mean predicted pLDDT scores for the ESMFold predicted structures. We focused on proteins with geNomad markers from generated sequences (sample size: n = 343; median value: 28) against random peptide sequences of the same lengths (sample size: n = 343; median value: 18). A sample generated protein is shown on the right. Two-sided Kolmogorov–Smirnov test: p value = 6.7 × 10−42. i Top 10 predicted functions of proteins derived from the generated sequences, as identified by phold. Source data are provided as a Source Data file.
Similar articles
-
Transformer model generated bacteriophage genomes are compositionally distinct from natural sequences.NAR Genom Bioinform. 2024 Sep 18;6(3):lqae129. doi: 10.1093/nargab/lqae129. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39296932 Free PMC article.
-
In silico characterization of DNA motifs with particular reference to promoters and terminators.Methods Mol Biol. 2009;502:113-29. doi: 10.1007/978-1-60327-565-1_8. Methods Mol Biol. 2009. PMID: 19082554
-
DeepPL: A deep-learning-based tool for the prediction of bacteriophage lifecycle.PLoS Comput Biol. 2024 Oct 17;20(10):e1012525. doi: 10.1371/journal.pcbi.1012525. eCollection 2024 Oct. PLoS Comput Biol. 2024. PMID: 39418300 Free PMC article.
-
A Bioinformatic Ecosystem for Bacteriophage Genomics: PhaMMSeqs, Phamerator, pdm_utils, PhagesDB, DEPhT, and PhamClust.Viruses. 2024 Aug 10;16(8):1278. doi: 10.3390/v16081278. Viruses. 2024. PMID: 39205252 Free PMC article. Review.
-
Analysis of the phage sequence space: the benefit of structured information.Virology. 2007 Sep 1;365(2):241-9. doi: 10.1016/j.virol.2007.03.047. Epub 2007 May 7. Virology. 2007. PMID: 17482656 Review.
Cited by
-
The design and engineering of synthetic genomes.Nat Rev Genet. 2024 Nov 6. doi: 10.1038/s41576-024-00786-y. Online ahead of print. Nat Rev Genet. 2024. PMID: 39506144 Review.
-
Protein Set Transformer: A protein-based genome language model to power high diversity viromics.Res Sq [Preprint]. 2024 Sep 23:rs.3.rs-4844047. doi: 10.21203/rs.3.rs-4844047/v1. Res Sq. 2024. PMID: 39399683 Free PMC article. Preprint.
-
Synthetic genomes unveil the effects of synonymous recoding.bioRxiv [Preprint]. 2024 Jun 16:2024.06.16.599206. doi: 10.1101/2024.06.16.599206. bioRxiv. 2024. PMID: 38915524 Free PMC article. Preprint.
-
Protein Set Transformer: A protein-based genome language model to power high diversity viromics.bioRxiv [Preprint]. 2024 Jul 29:2024.07.26.605391. doi: 10.1101/2024.07.26.605391. bioRxiv. 2024. PMID: 39131363 Free PMC article. Preprint.
-
Evaluating the representational power of pre-trained DNA language models for regulatory genomics.bioRxiv [Preprint]. 2024 Sep 25:2024.02.29.582810. doi: 10.1101/2024.02.29.582810. bioRxiv. 2024. PMID: 38464101 Free PMC article. Preprint.
References
-
- Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: pre-training of deep bidirectional transformers for language understanding. Preprint at arXiv10.48550/arXiv.1810.04805 (2018).
-
- Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst.33, 1877–1901 (2020).
-
- Benegas, G., Ye, C., Albors, C., Li, J. C. & Song, Y. S. Genomic language models: opportunities and challenges. Preprint at arXiv10.48550/arXiv.2407.11435 (2024).
-
- Dalla-Torre, H. et al. The nucleotide transformer: building and evaluating robust foundation models for human genomics. bioRxiv10.1101/2023.01.11.523679 (2023).
MeSH terms
LinkOut - more resources
Full Text Sources
