

A Rigorous Framework for Calculating Protein-Protein Binding Affinities in Membranes
- PMID: 38091976
- PMCID: PMC11145395
- DOI: 10.1021/acs.jctc.3c00941
A Rigorous Framework for Calculating Protein-Protein Binding Affinities in Membranes
Abstract
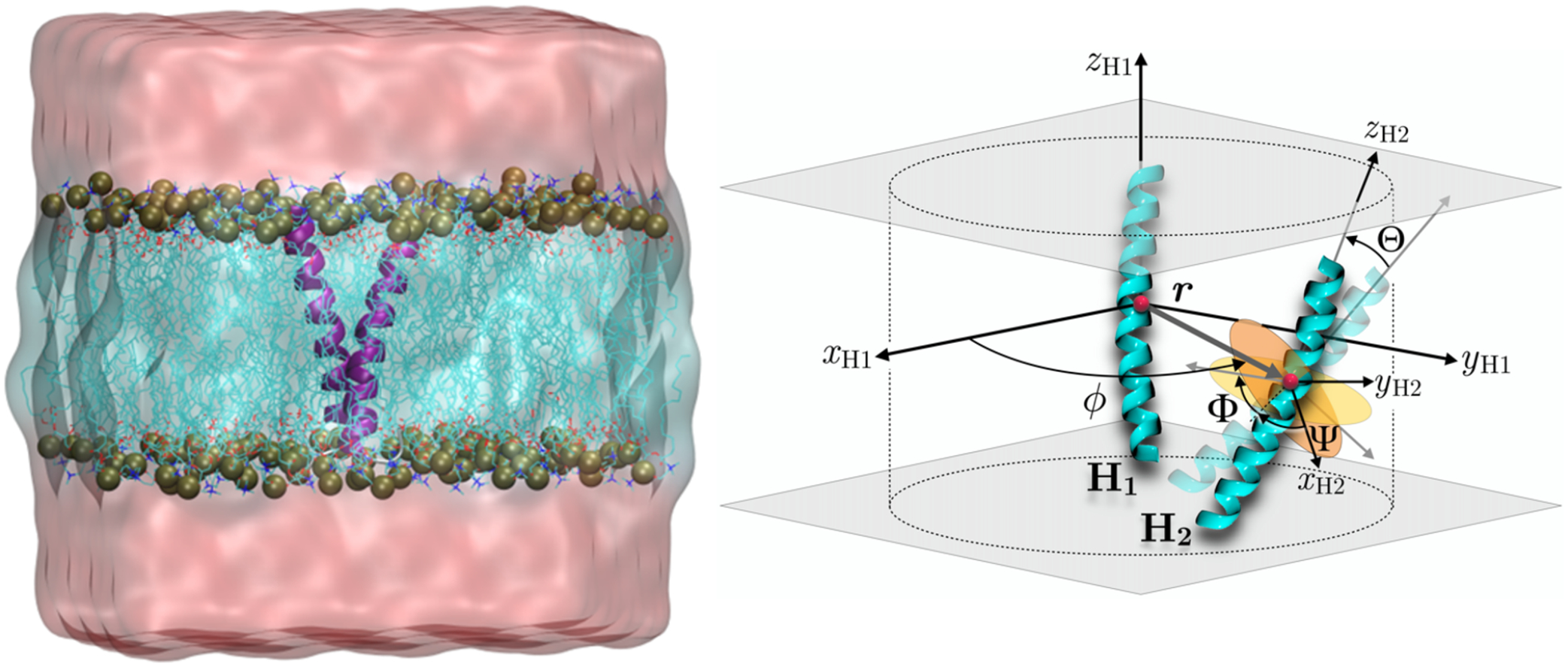
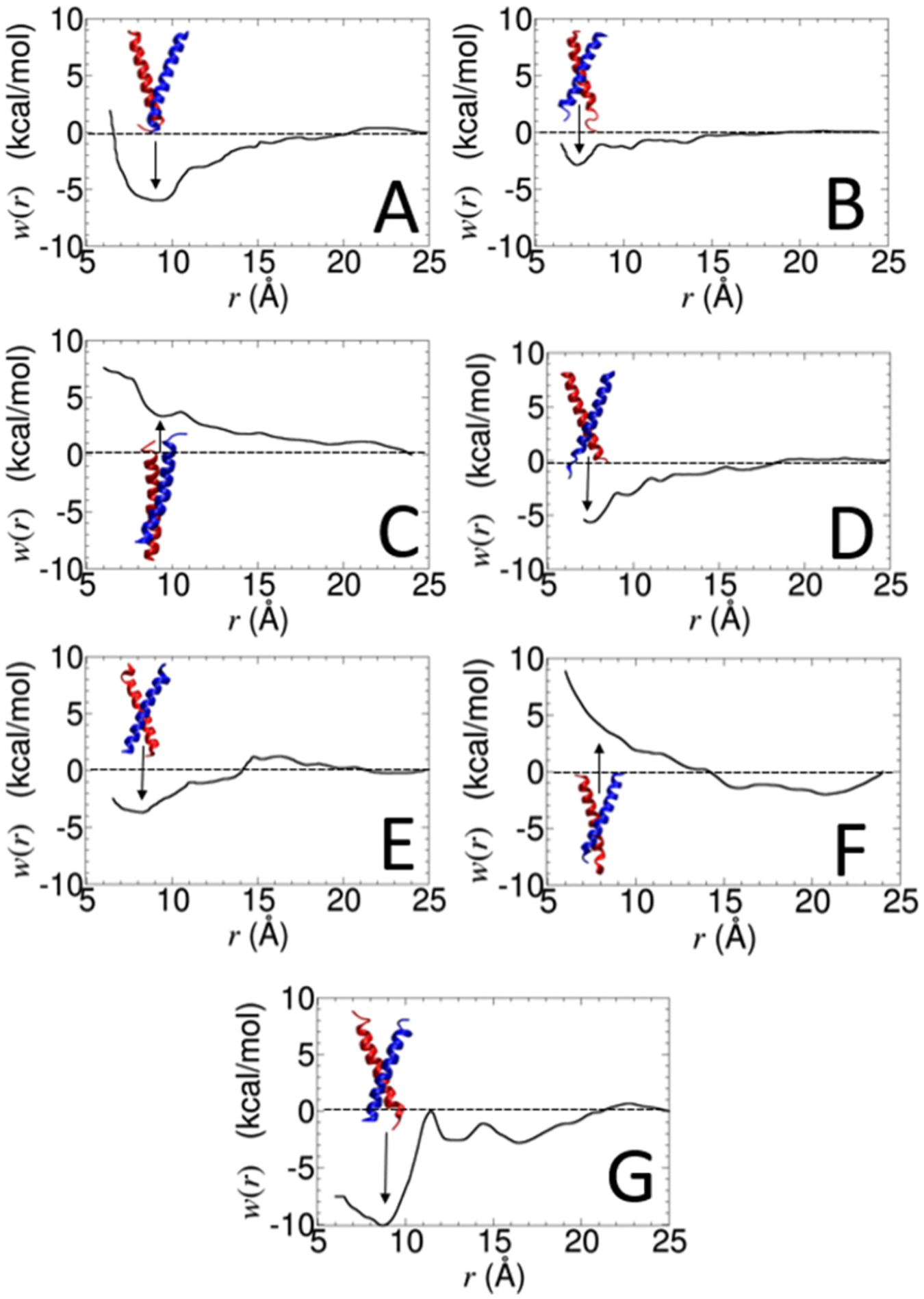
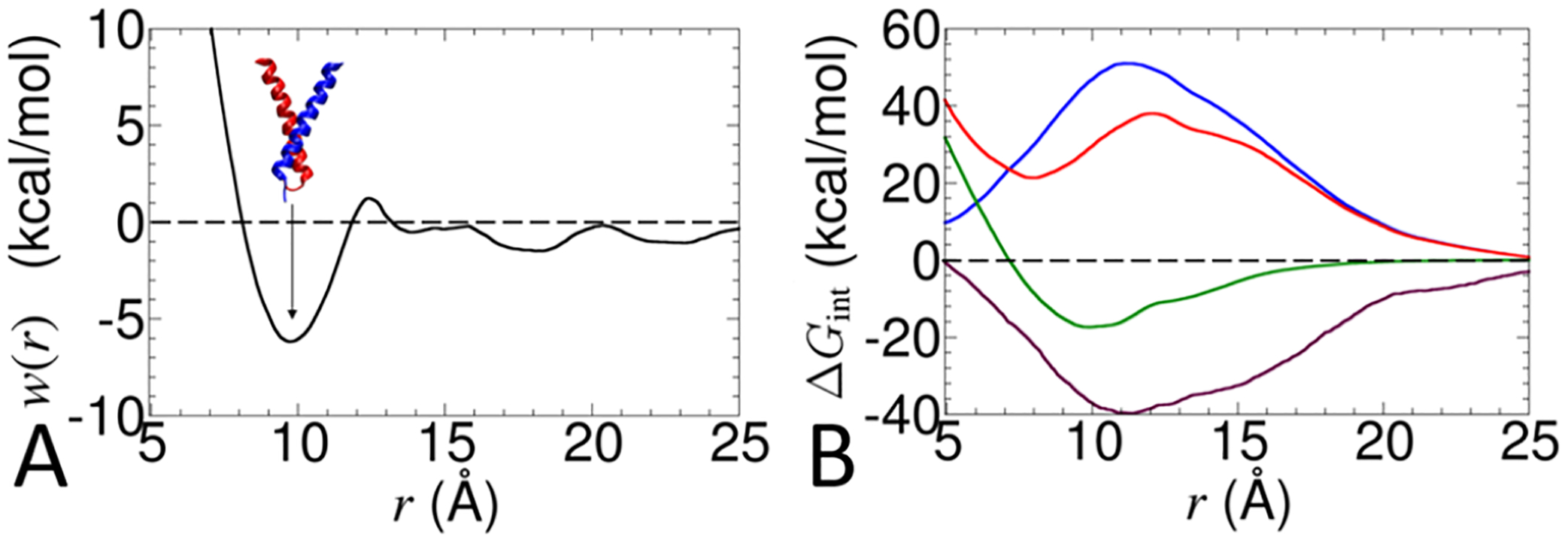
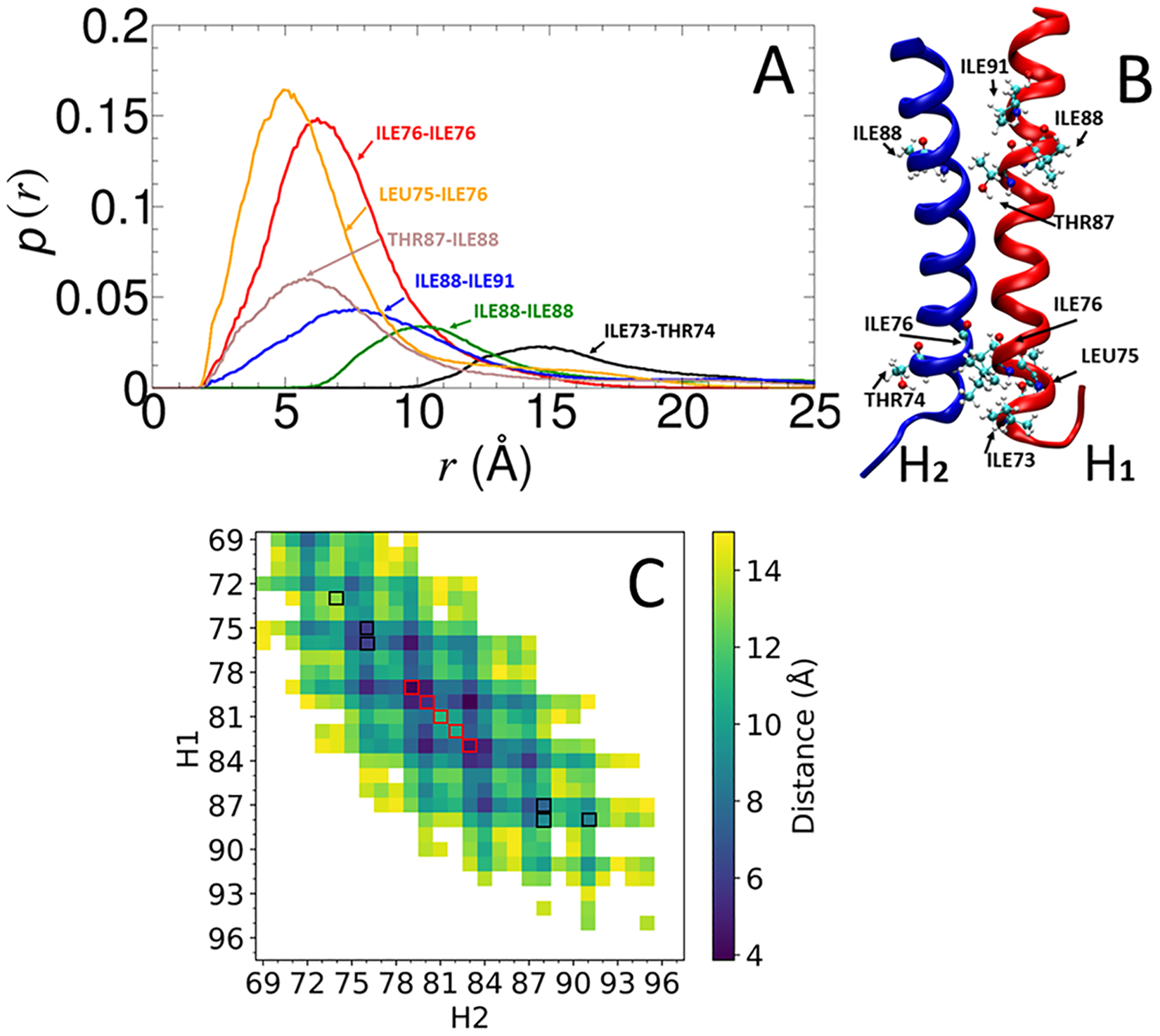
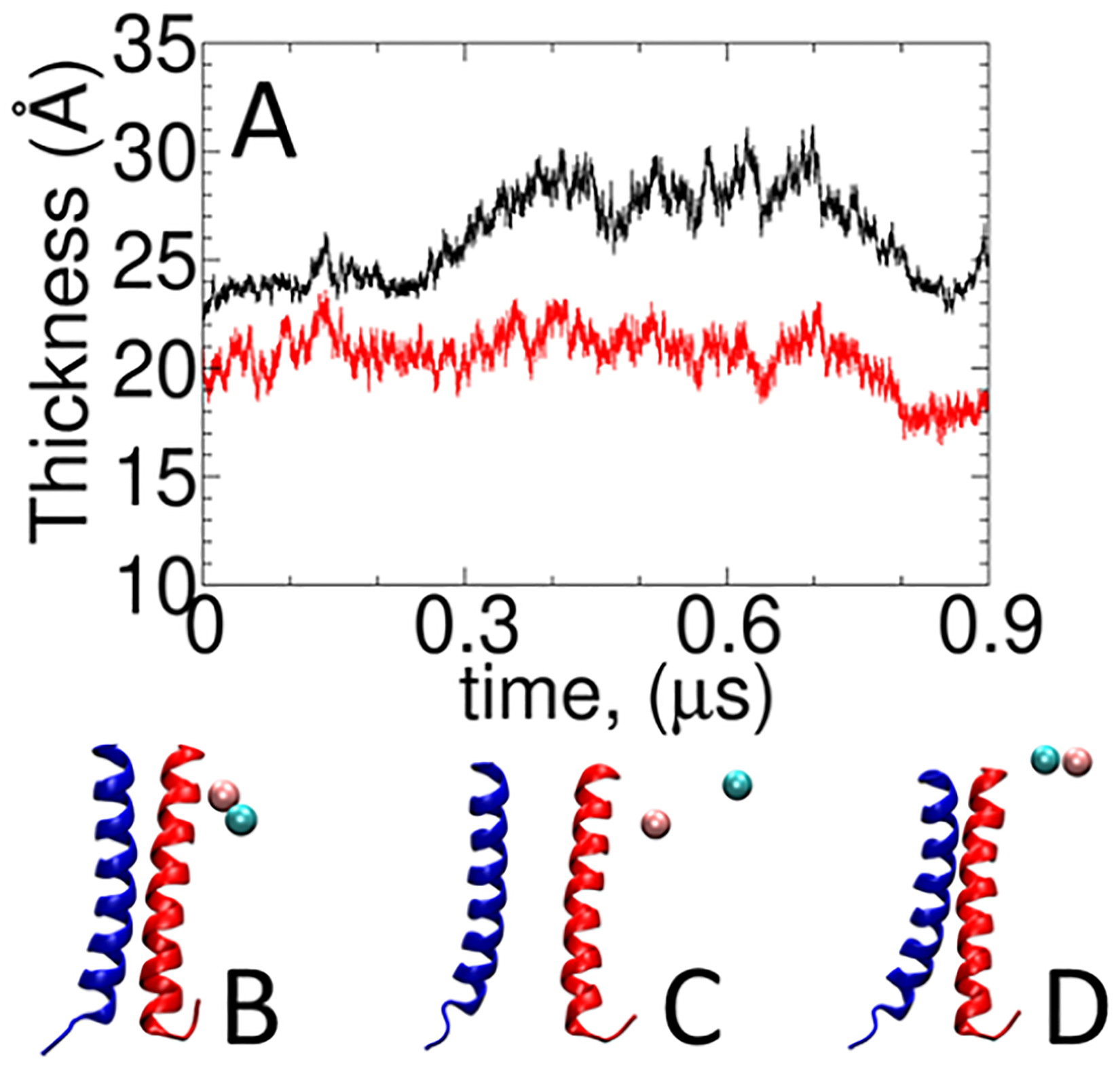
Calculating the binding free energy of integral transmembrane (TM) proteins is crucial for understanding the mechanisms by which they recognize one another and reversibly associate. The glycophorin A (GpA) homodimer, composed of two α-helical segments, has long served as a model system for studying TM protein reversible association. The present work establishes a methodological framework for calculating the binding affinity of the GpA homodimer in the heterogeneous environment of a membrane. Our investigation carefully considered a variety of protocols, including the appropriate choice of the force field, rigorous standardization reflecting the experimental conditions, sampling algorithm, anisotropic environment, and collective variables, to accurately describe GpA dimerization via molecular dynamics-based approaches. Specifically, two strategies were explored: (i) an unrestrained potential mean force (PMF) calculation, which merely enhances sampling along the separation of the two binding partners without any restraint, and (ii) a so-called "geometrical route", whereby the α-helices are progressively separated with imposed restraints on their orientational, positional, and conformational degrees of freedom to accelerate convergence. Our simulations reveal that the simplified, unrestrained PMF approach is inadequate for the description of GpA dimerization. Instead, the geometrical route, tailored specifically to GpA in a membrane environment, yields excellent agreement with experimental data within a reasonable computational time. A dimerization free energy of -10.7 kcal/mol is obtained, in fairly good agreement with available experimental data. The geometrical route further helps elucidate how environmental forces drive association before helical interactions stabilize it. Our simulations also brought to light a distinct, long-lived spatial arrangement that potentially serves as an intermediate state during dimer formation. The methodological advances in the generalized geometrical route provide a powerful tool for accurate and efficient binding-affinity calculations of intricate TM protein complexes in inhomogeneous environments.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

Similar articles
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Defining the optimum strategy for identifying adults and children with coeliac disease: systematic review and economic modelling.Health Technol Assess. 2022 Oct;26(44):1-310. doi: 10.3310/ZUCE8371. Health Technol Assess. 2022. PMID: 36321689 Free PMC article.
-
The effectiveness of school-based family asthma educational programs on the quality of life and number of asthma exacerbations of children aged five to 18 years diagnosed with asthma: a systematic review protocol.JBI Database System Rev Implement Rep. 2015 Oct;13(10):69-81. doi: 10.11124/jbisrir-2015-2335. JBI Database System Rev Implement Rep. 2015. PMID: 26571284
-
Using Experience Sampling Methodology to Capture Disclosure Opportunities for Autistic Adults.Autism Adulthood. 2023 Dec 1;5(4):389-400. doi: 10.1089/aut.2022.0090. Epub 2023 Dec 12. Autism Adulthood. 2023. PMID: 38116059 Free PMC article.
-
Impact of residual disease as a prognostic factor for survival in women with advanced epithelial ovarian cancer after primary surgery.Cochrane Database Syst Rev. 2022 Sep 26;9(9):CD015048. doi: 10.1002/14651858.CD015048.pub2. Cochrane Database Syst Rev. 2022. PMID: 36161421 Free PMC article. Review.
Cited by
-
Machine Learning Derived Collective Variables for the Study of Protein Homodimerization in Membrane.J Chem Theory Comput. 2024 Jul 9;20(13):5774-5783. doi: 10.1021/acs.jctc.4c00454. Epub 2024 Jun 25. J Chem Theory Comput. 2024. PMID: 38918177
References
-
- Adams PD; Engelman DM; nger AT Improved prediction for the structure of the dimeric transmembrane domain of glycophorin A obtained through global searching. Proteins 1996, 26, 257–261. - PubMed
-
- Popot JL; Engelman DM Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem 2000, 69, 881–922. - PubMed
-
- Booth PJ; Templer RH; Meijberg W; Allen SJ; Curran AR; Lorch M In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol 2001, 36, 501–603. - PubMed
-
- Arkin IT Structural aspects of oligomerization taking place between the transmembrane α-helices of bitopic membrane proteins. Biochim. Biophys. Acta - Biomembr 2002, 1565, 347–363. - PubMed
-
- Chin C-N; von Heijne G; de Gier J-WL Membrane proteins: shaping up. Trends Biochem. Sci 2002, 27, 231–234. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources