

Integrative multi-omic cancer profiling reveals DNA methylation patterns associated with therapeutic vulnerability and cell-of-origin
- PMID: 37582362
- PMCID: PMC11613269
- DOI: 10.1016/j.ccell.2023.07.013
Integrative multi-omic cancer profiling reveals DNA methylation patterns associated with therapeutic vulnerability and cell-of-origin
Abstract
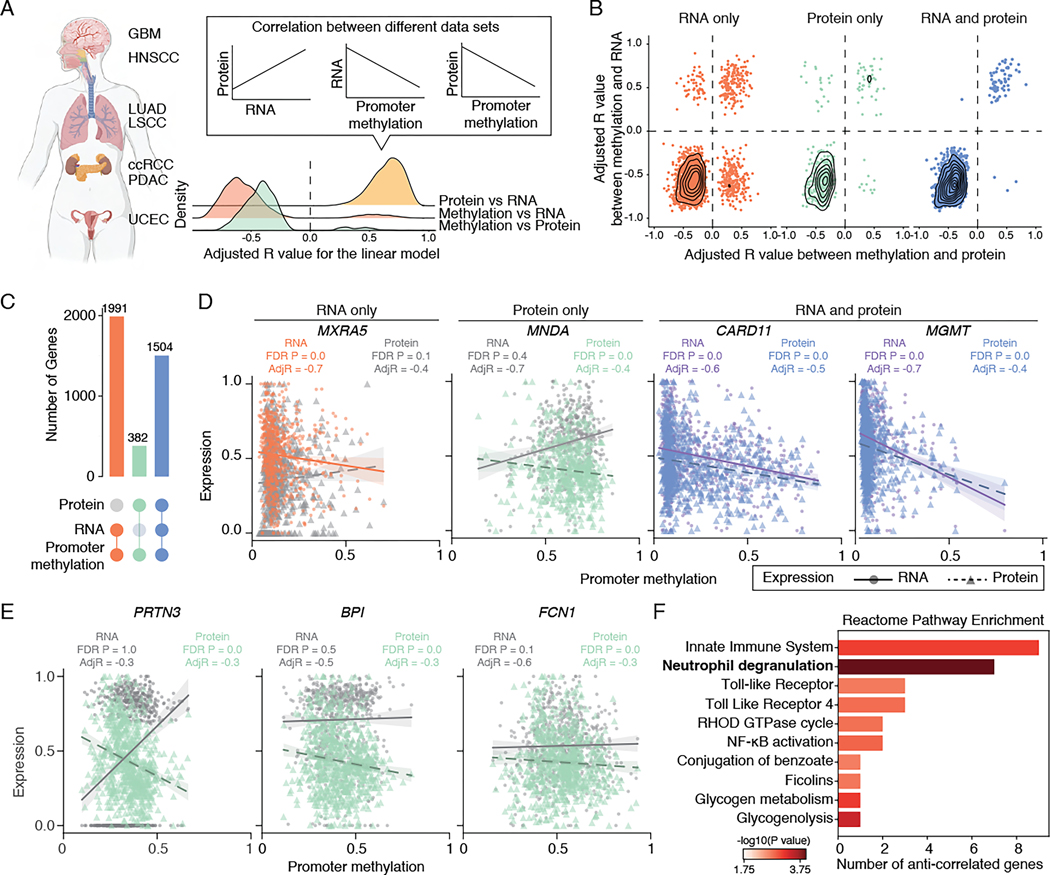
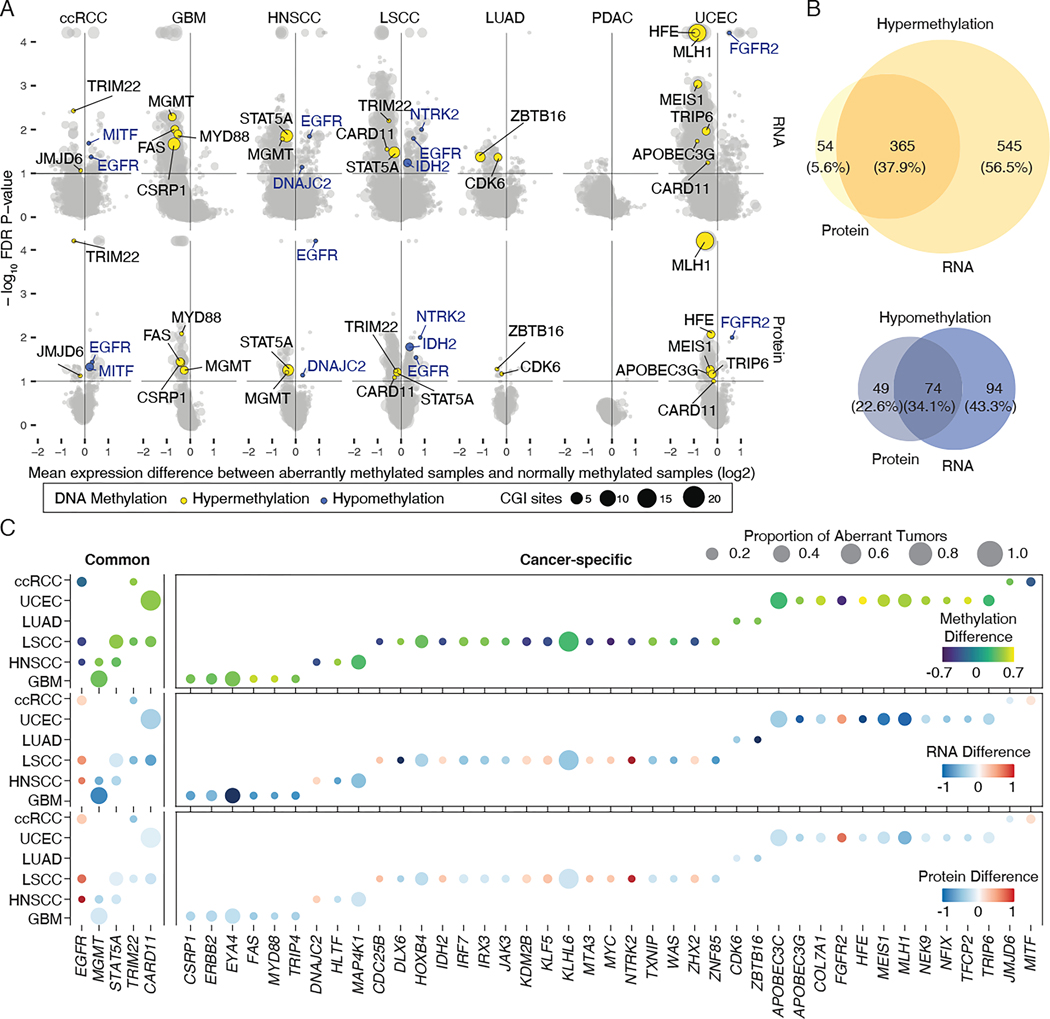
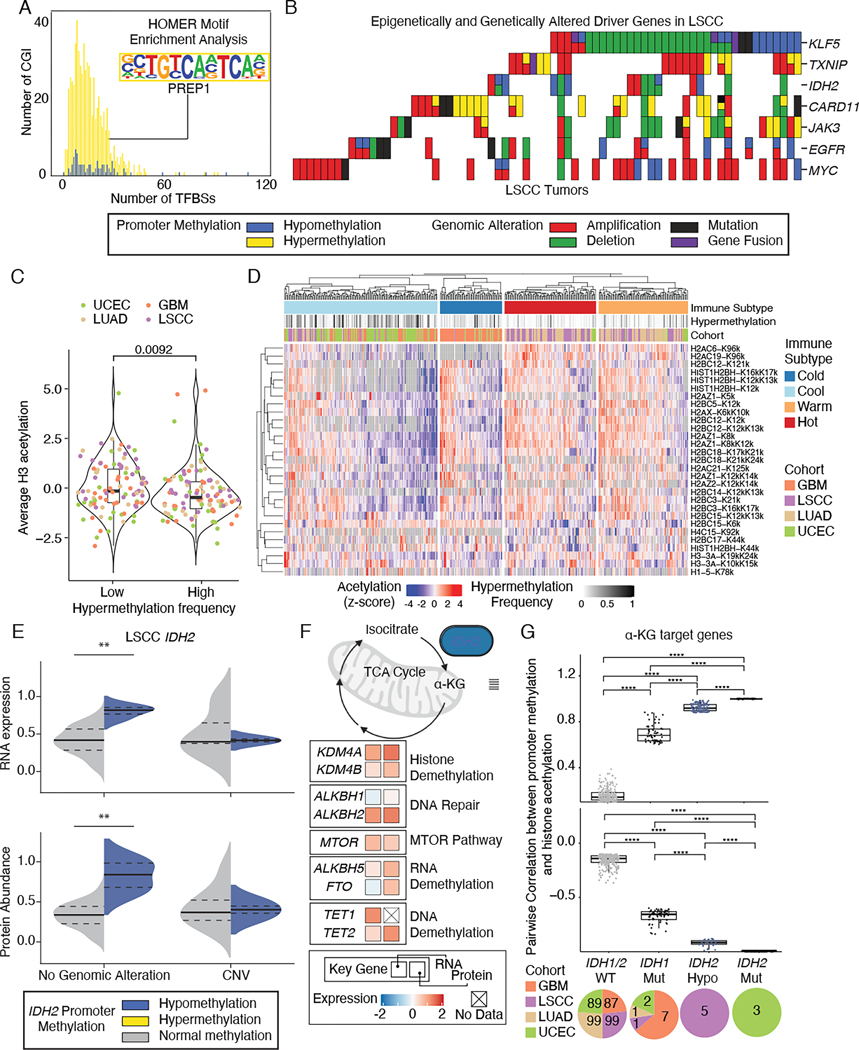
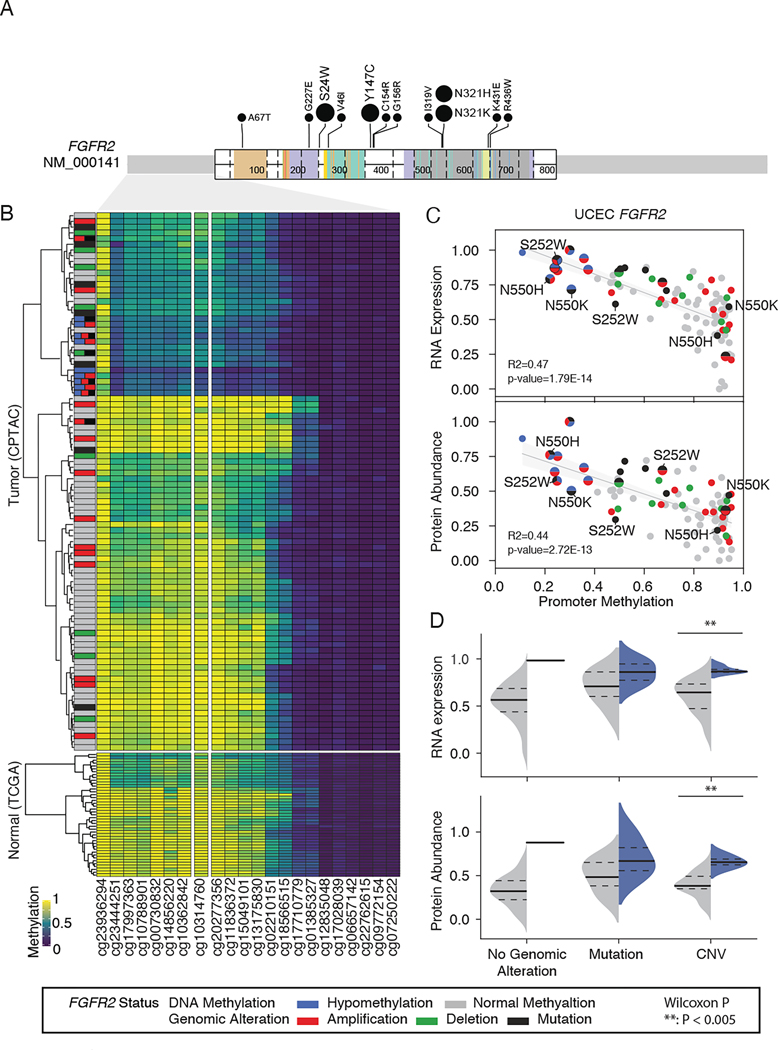
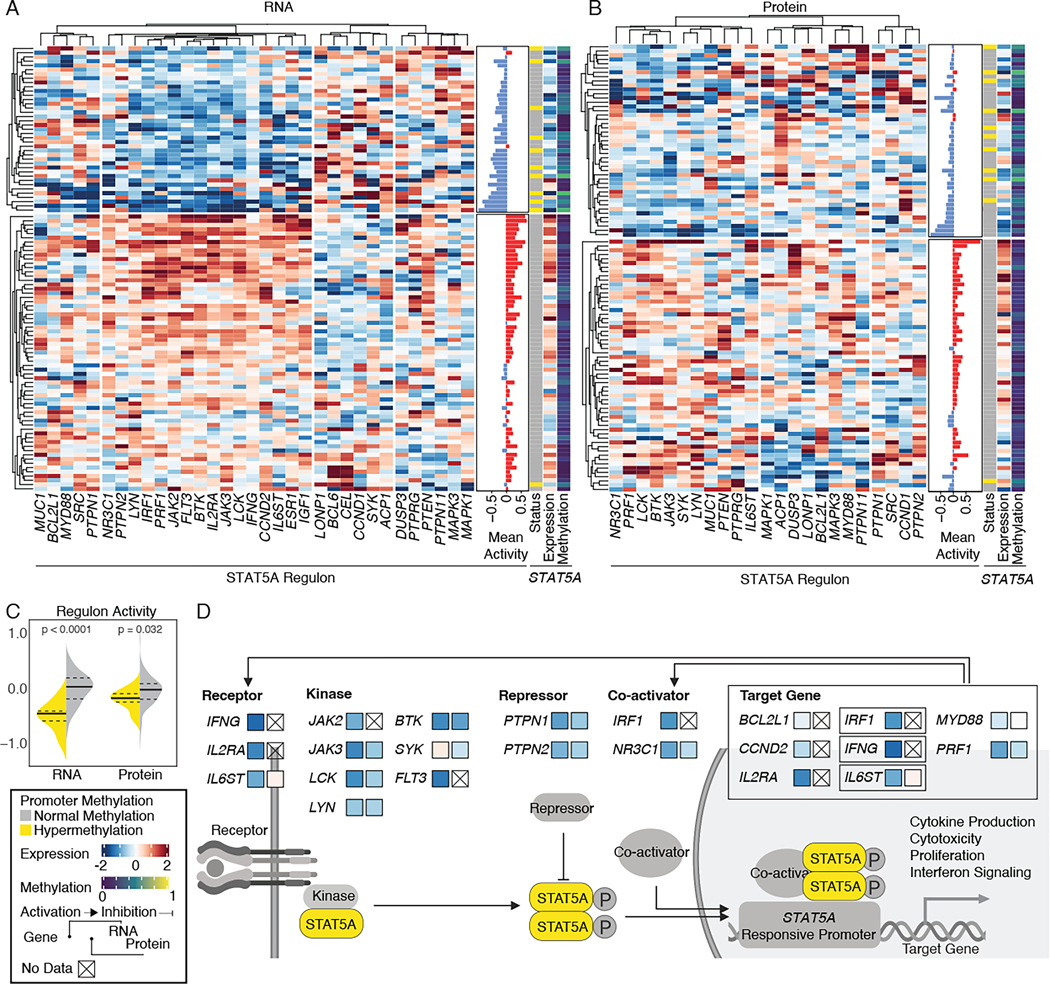
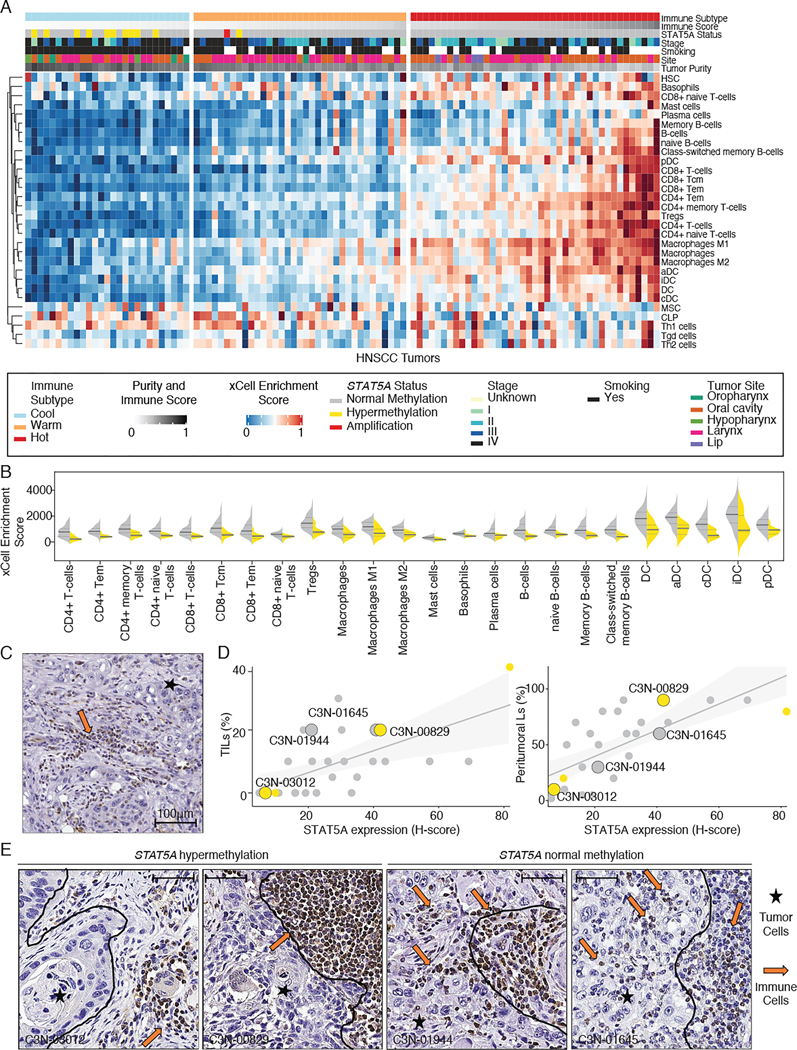
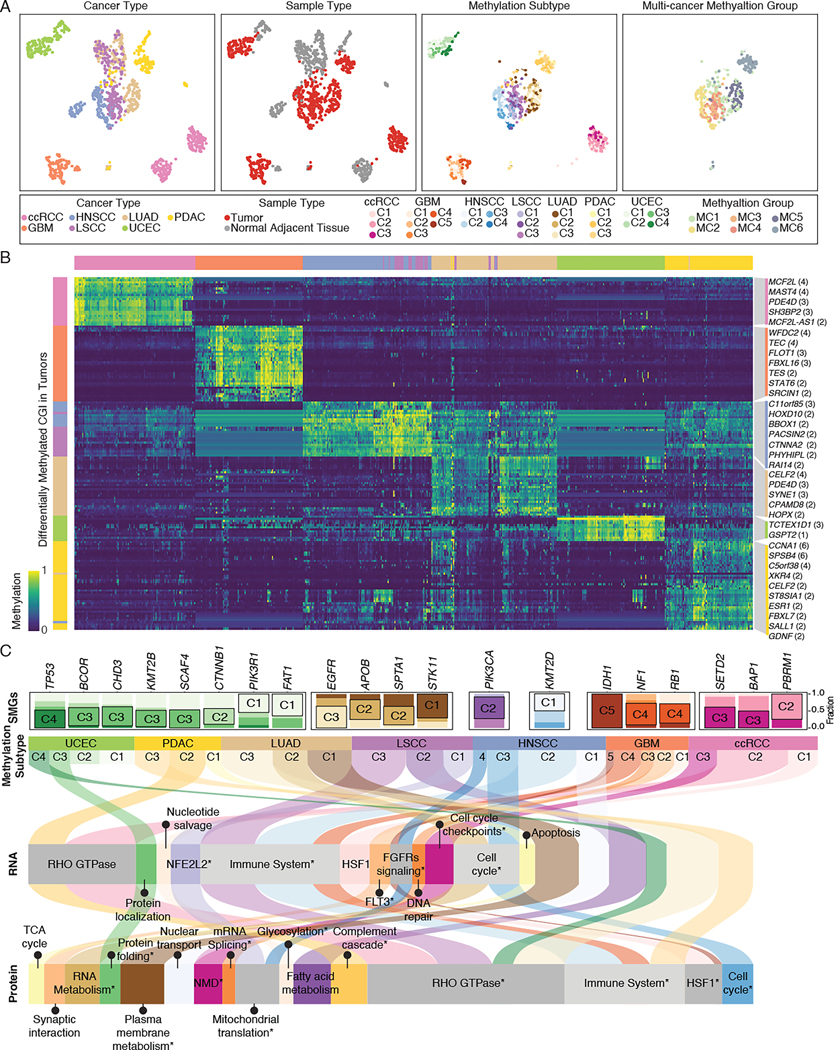
DNA methylation plays a critical role in establishing and maintaining cellular identity. However, it is frequently dysregulated during tumor development and is closely intertwined with other genetic alterations. Here, we leveraged multi-omic profiling of 687 tumors and matched non-involved adjacent tissues from the kidney, brain, pancreas, lung, head and neck, and endometrium to identify aberrant methylation associated with RNA and protein abundance changes and build a Pan-Cancer catalog. We uncovered lineage-specific epigenetic drivers including hypomethylated FGFR2 in endometrial cancer. We showed that hypermethylated STAT5A is associated with pervasive regulon downregulation and immune cell depletion, suggesting that epigenetic regulation of STAT5A expression constitutes a molecular switch for immunosuppression in squamous tumors. We further demonstrated that methylation subtype-enrichment information can explain cell-of-origin, intra-tumor heterogeneity, and tumor phenotypes. Overall, we identified cis-acting DNA methylation events that drive transcriptional and translational changes, shedding light on the tumor's epigenetic landscape and the role of its cell-of-origin.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Similar articles
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Identifying genes associated with disease outcomes using joint sparse canonical correlation analysis-An application in renal clear cell carcinoma.Genet Epidemiol. 2024 Dec;48(8):414-432. doi: 10.1002/gepi.22566. Epub 2024 May 15. Genet Epidemiol. 2024. PMID: 38751238 Free PMC article.
-
Enabling Systemic Identification and Functionality Profiling for Cdc42 Homeostatic Modulators.bioRxiv [Preprint]. 2024 Jan 8:2024.01.05.574351. doi: 10.1101/2024.01.05.574351. bioRxiv. 2024. Update in: Commun Chem. 2024 Nov 19;7(1):271. doi: 10.1038/s42004-024-01352-7 PMID: 38260445 Free PMC article. Updated. Preprint.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Metformin for endometrial hyperplasia.Cochrane Database Syst Rev. 2024 May 2;5(5):CD012214. doi: 10.1002/14651858.CD012214.pub3. Cochrane Database Syst Rev. 2024. PMID: 38695827 Review.
Cited by
-
DNA Methylation as a Molecular Mechanism of Carcinogenesis in World Trade Center Dust Exposure: Insights from a Structured Literature Review.Biomolecules. 2024 Oct 15;14(10):1302. doi: 10.3390/biom14101302. Biomolecules. 2024. PMID: 39456235 Free PMC article. Review.
-
Integrated profiling identifies DXS253E as a potential prognostic marker in colorectal cancer.Cancer Cell Int. 2024 Jun 18;24(1):213. doi: 10.1186/s12935-024-03403-4. Cancer Cell Int. 2024. PMID: 38890691 Free PMC article.
-
Tissue of origin prediction for cancer of unknown primary using a targeted methylation sequencing panel.Clin Epigenetics. 2024 Feb 9;16(1):25. doi: 10.1186/s13148-024-01638-6. Clin Epigenetics. 2024. PMID: 38336771 Free PMC article.
-
Regulatory effect of N6-methyladenosine on tumor angiogenesis.Front Immunol. 2024 Sep 4;15:1453774. doi: 10.3389/fimmu.2024.1453774. eCollection 2024. Front Immunol. 2024. PMID: 39295872 Free PMC article. Review.
-
The DNA methylome of pediatric brain tumors appears shaped by structural variation and predicts survival.Nat Commun. 2024 Aug 8;15(1):6775. doi: 10.1038/s41467-024-51276-y. Nat Commun. 2024. PMID: 39117669 Free PMC article.
References
-
- Feinberg AP, Ohlsson R, and Henikoff S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33. - PubMed
-
- Ehrlich M. (2002). DNA methylation in cancer: too much, but also too little. Oncogene 21, 5400–5413. - PubMed
-
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CPE, van Dijk CM, Tollenaar RA, et al. (2012). Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina--associated domains. Nat. Genet. 44, 40. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- U24 CA210954/CA/NCI NIH HHS/United States
- U01 CA214116/CA/NCI NIH HHS/United States
- U24 CA210985/CA/NCI NIH HHS/United States
- U24 CA210967/CA/NCI NIH HHS/United States
- U24 CA210986/CA/NCI NIH HHS/United States
- HHSN261201500003C/CA/NCI NIH HHS/United States
- U24 CA210972/CA/NCI NIH HHS/United States
- U24 CA210955/CA/NCI NIH HHS/United States
- U24 CA210993/CA/NCI NIH HHS/United States
- HHSN261201500003I/CA/NCI NIH HHS/United States
- U24 CA210979/CA/NCI NIH HHS/United States
- U24 CA271012/CA/NCI NIH HHS/United States
- U01 CA214114/CA/NCI NIH HHS/United States
- U01 CA214125/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
