

Design, construction, and in vivo augmentation of a complex gut microbiome
- PMID: 36070752
- PMCID: PMC9691261
- DOI: 10.1016/j.cell.2022.08.003
Design, construction, and in vivo augmentation of a complex gut microbiome
Abstract
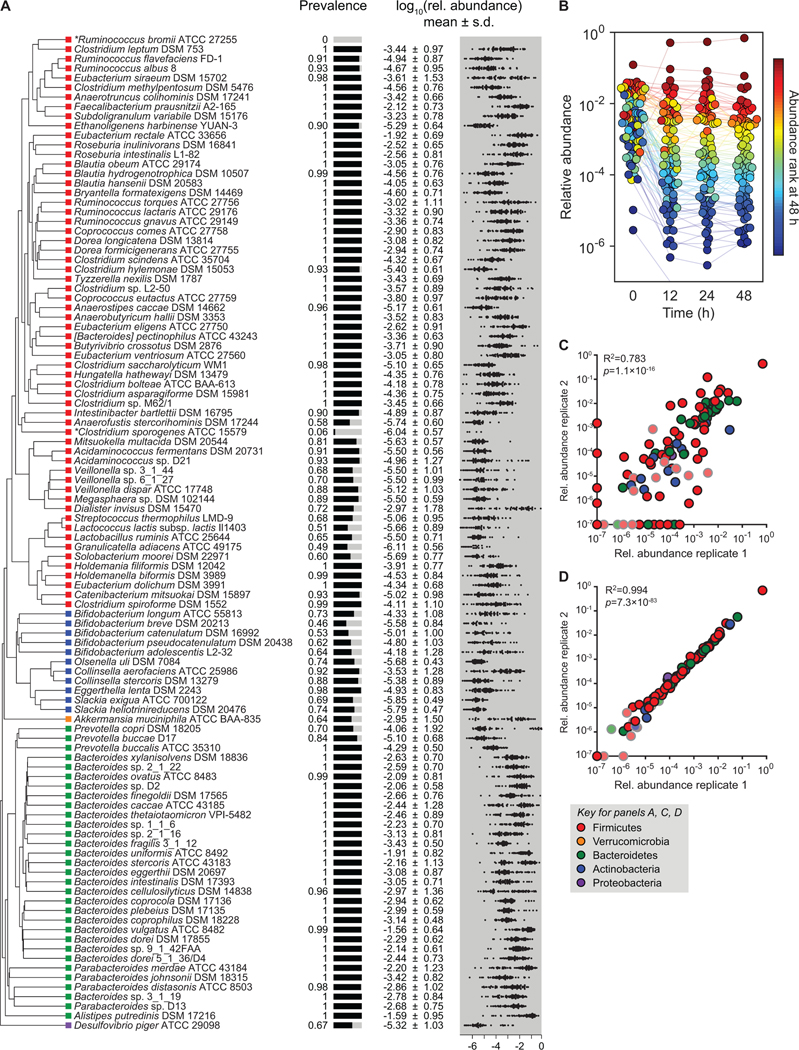
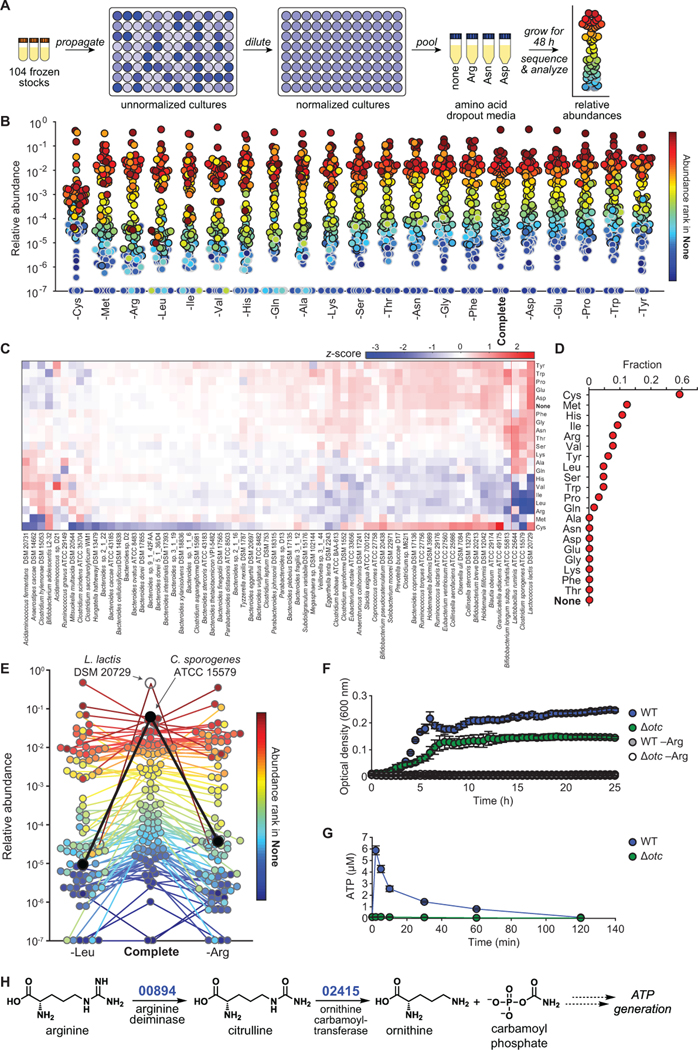
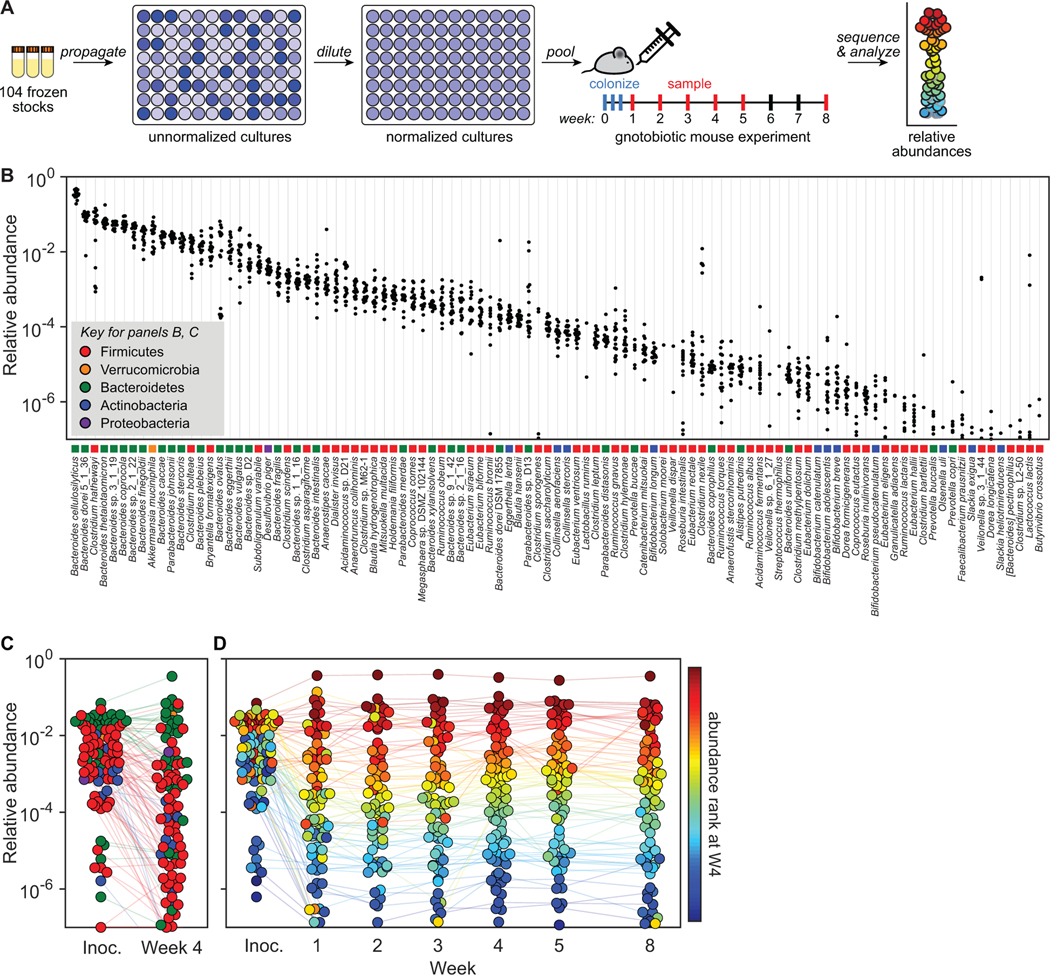
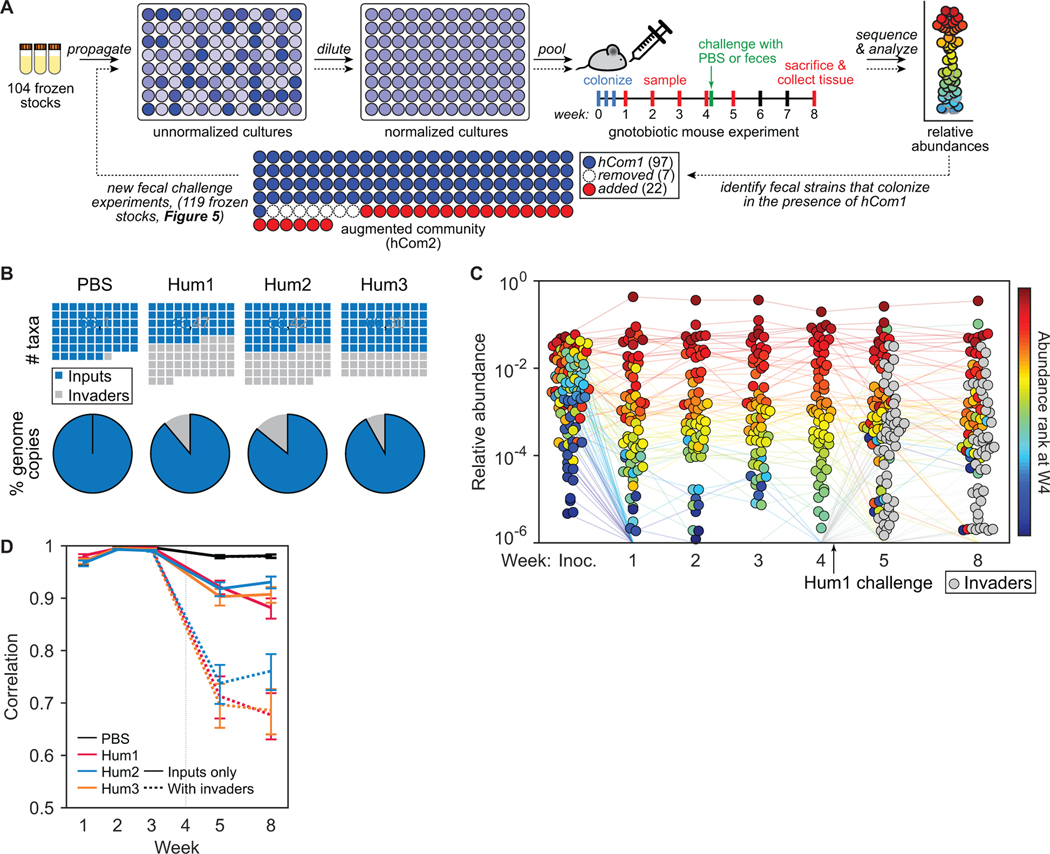
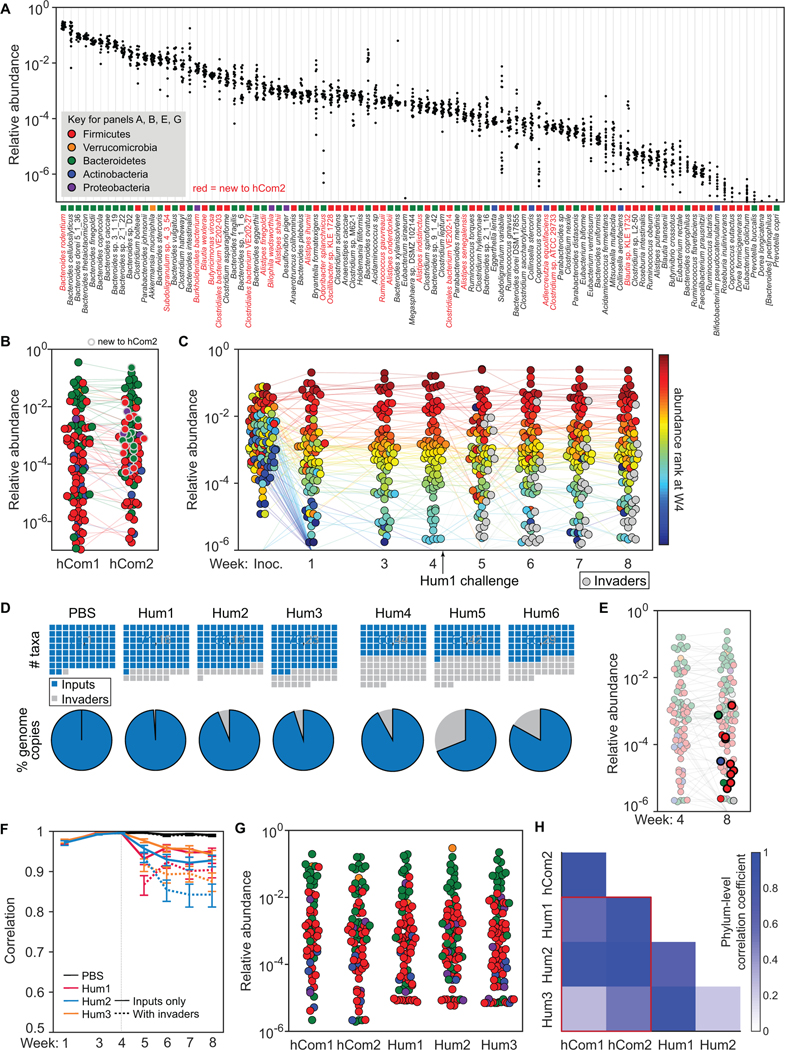
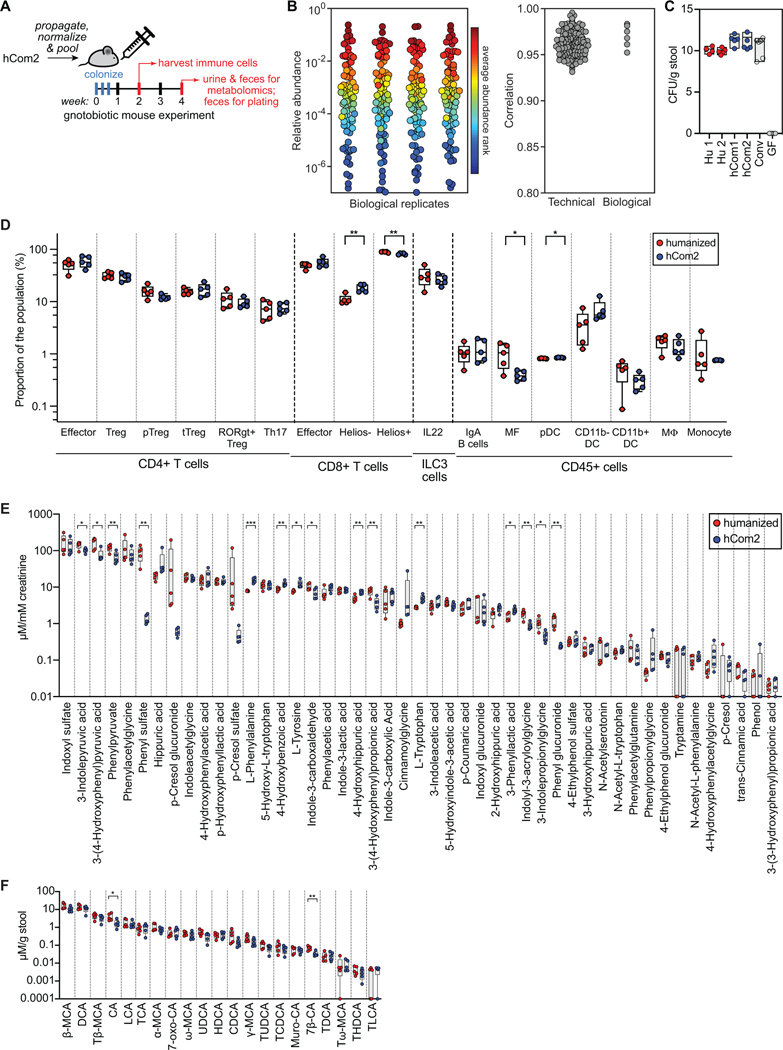
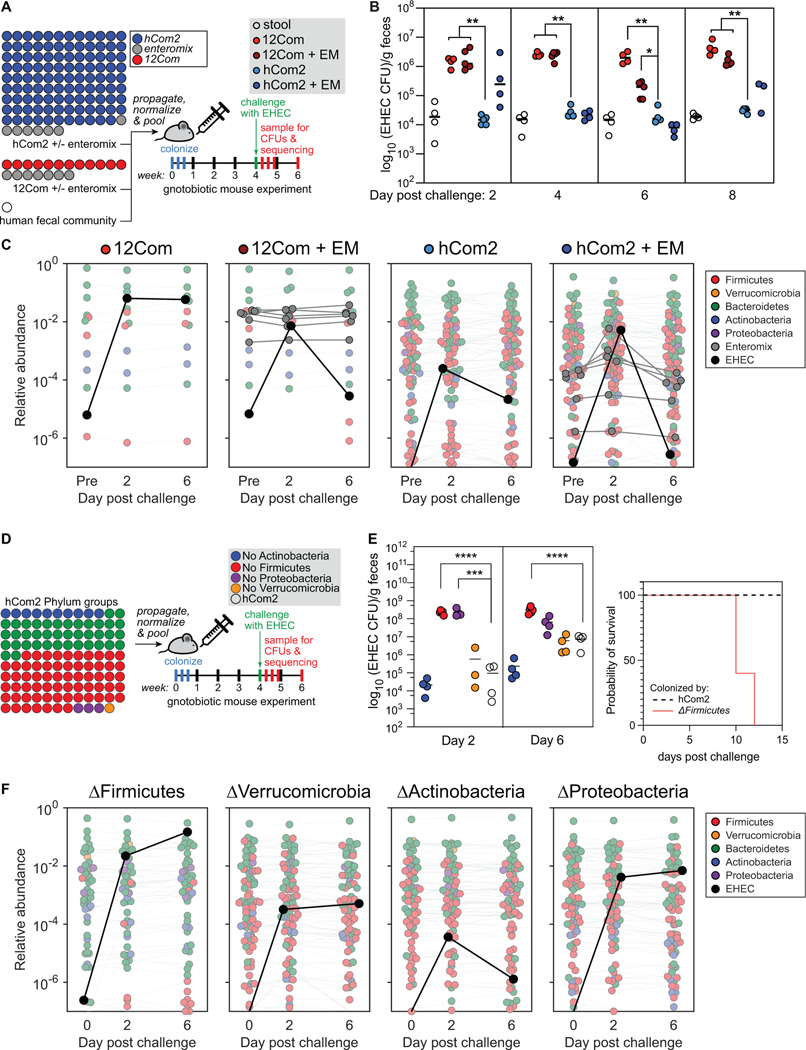
Efforts to model the human gut microbiome in mice have led to important insights into the mechanisms of host-microbe interactions. However, the model communities studied to date have been defined or complex, but not both, limiting their utility. Here, we construct and characterize in vitro a defined community of 104 bacterial species composed of the most common taxa from the human gut microbiota (hCom1). We then used an iterative experimental process to fill open niches: germ-free mice were colonized with hCom1 and then challenged with a human fecal sample. We identified new species that engrafted following fecal challenge and added them to hCom1, yielding hCom2. In gnotobiotic mice, hCom2 exhibited increased stability to fecal challenge and robust colonization resistance against pathogenic Escherichia coli. Mice colonized by either hCom2 or a human fecal community are phenotypically similar, suggesting that this consortium will enable a mechanistic interrogation of species and genes on microbiome-associated phenotypes.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Stanford University and the Chan Zuckerberg Biohub have patents pending for microbiome technologies on which the authors are co-inventors. M.A.F. is a co-founder and director of Federation Bio and Kelonia, a co-founder of Revolution Medicines, and a member of the scientific advisory boards of NGM Bio and Zymergen. A.G.C. and K.N. have been paid consultants to Federation Bio. A.R.B. has been an employee of Federation Bio.
Figures
Similar articles
-
Microbiome Resilience despite a Profound Loss of Minority Microbiota following Clindamycin Challenge in Humanized Gnotobiotic Mice.Microbiol Spectr. 2022 Feb 23;10(1):e0196021. doi: 10.1128/spectrum.01960-21. Epub 2022 Jan 12. Microbiol Spectr. 2022. PMID: 35019780 Free PMC article.
-
Postnatal colonization with human "infant-type" Bifidobacterium species alters behavior of adult gnotobiotic mice.PLoS One. 2018 May 15;13(5):e0196510. doi: 10.1371/journal.pone.0196510. eCollection 2018. PLoS One. 2018. PMID: 29763437 Free PMC article.
-
Engraftment of aging-related human gut microbiota and the effect of a seven-species consortium in a pre-clinical model.Gut Microbes. 2023 Dec;15(2):2282796. doi: 10.1080/19490976.2023.2282796. Epub 2023 Nov 27. Gut Microbes. 2023. PMID: 38010168 Free PMC article.
-
Strategies to Dissect Host-Microbial Immune Interactions That Determine Mucosal Homeostasis vs. Intestinal Inflammation in Gnotobiotic Mice.Front Immunol. 2020 Feb 18;11:214. doi: 10.3389/fimmu.2020.00214. eCollection 2020. Front Immunol. 2020. PMID: 32133003 Free PMC article. Review.
-
Functional roles and metabolic niches in the honey bee gut microbiota.Curr Opin Microbiol. 2018 Jun;43:69-76. doi: 10.1016/j.mib.2017.12.009. Epub 2018 Jan 12. Curr Opin Microbiol. 2018. PMID: 29309997 Review.
Cited by
-
StrainR2 accurately deconvolutes strain-level abundances in synthetic microbial communities.bioRxiv [Preprint]. 2024 Aug 9:2024.08.08.607172. doi: 10.1101/2024.08.08.607172. bioRxiv. 2024. PMID: 39149354 Free PMC article. Preprint.
-
Microbiome influencers of checkpoint blockade-associated toxicity.J Exp Med. 2023 Mar 6;220(3):e20220948. doi: 10.1084/jem.20220948. Epub 2023 Jan 9. J Exp Med. 2023. PMID: 36622383 Free PMC article.
-
Integrated multi-omic analyses provide insight into colon adenoma susceptibility modulation by the gut microbiota.mSystems. 2023 Aug 31;8(4):e0015123. doi: 10.1128/msystems.00151-23. Epub 2023 Jul 17. mSystems. 2023. PMID: 37458451 Free PMC article.
-
Application of MALDI-TOF MS for enumerating bacterial constituents of defined consortia.Appl Microbiol Biotechnol. 2023 Jun;107(12):4069-4077. doi: 10.1007/s00253-023-12558-5. Epub 2023 May 6. Appl Microbiol Biotechnol. 2023. PMID: 37148337 Free PMC article.
-
Strain dropouts reveal interactions that govern the metabolic output of the gut microbiome.Cell. 2023 Jun 22;186(13):2839-2852.e21. doi: 10.1016/j.cell.2023.05.037. Cell. 2023. PMID: 37352836 Free PMC article.
References
-
- Blasche S, Kim Y, Oliveira AP, and Patil KR (2017). Model microbial communities for ecosystems biology. Current Opinion in Systems Biology 6, 51–57.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
