

simplifyEnrichment: A Bioconductor Package for Clustering and Visualizing Functional Enrichment Results
- PMID: 35680096
- PMCID: PMC10373083
- DOI: 10.1016/j.gpb.2022.04.008
simplifyEnrichment: A Bioconductor Package for Clustering and Visualizing Functional Enrichment Results
Abstract
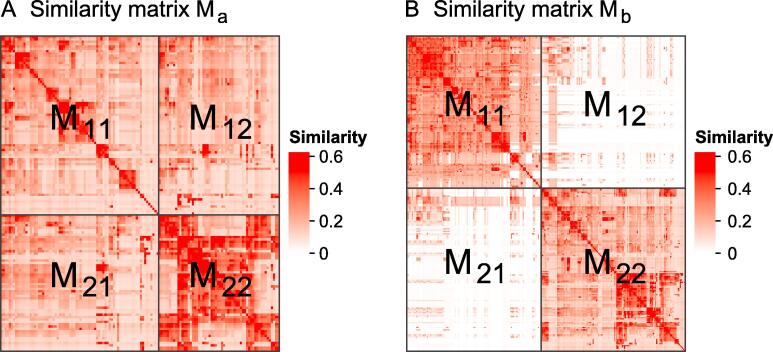
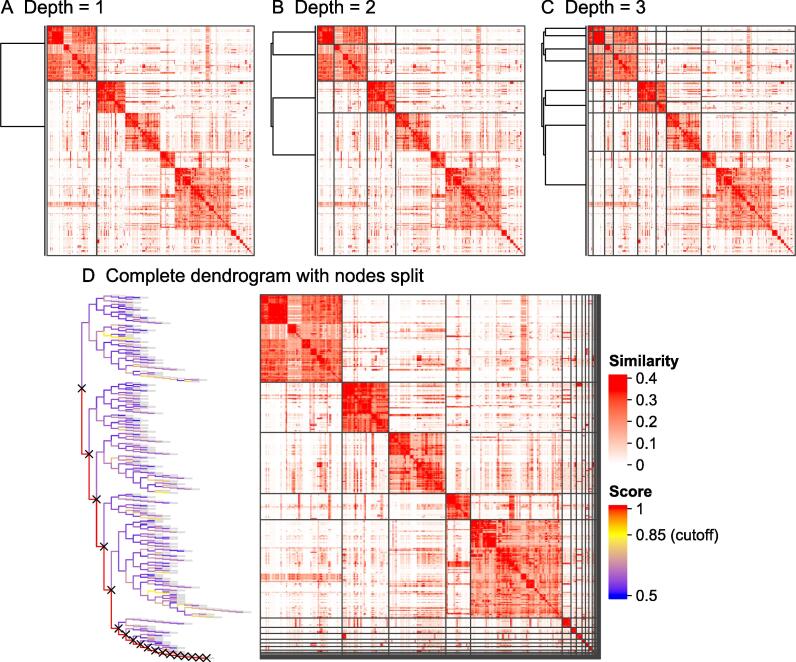
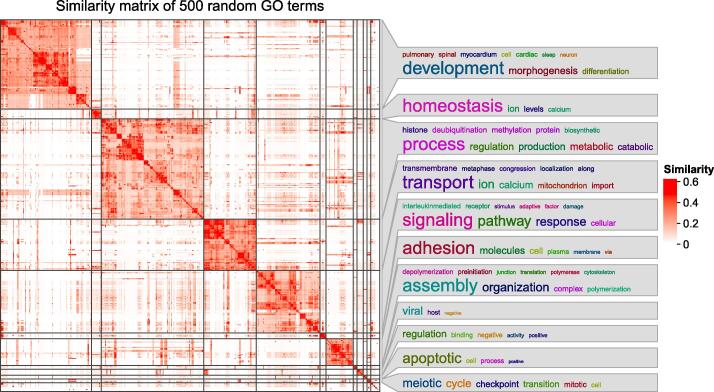
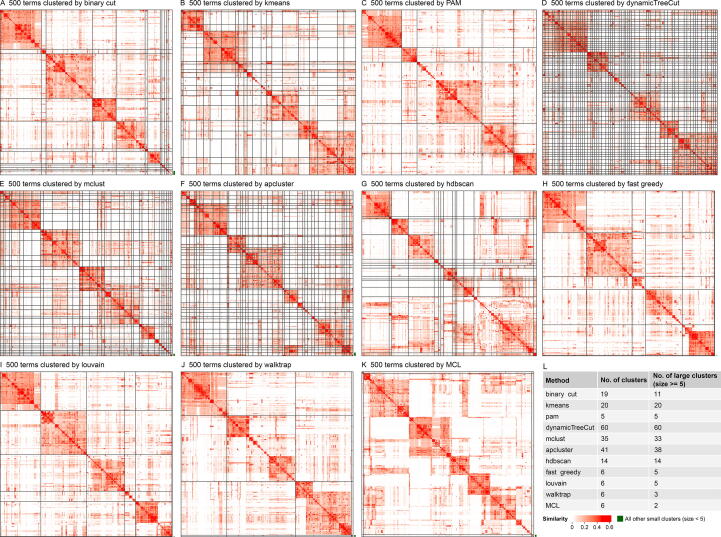
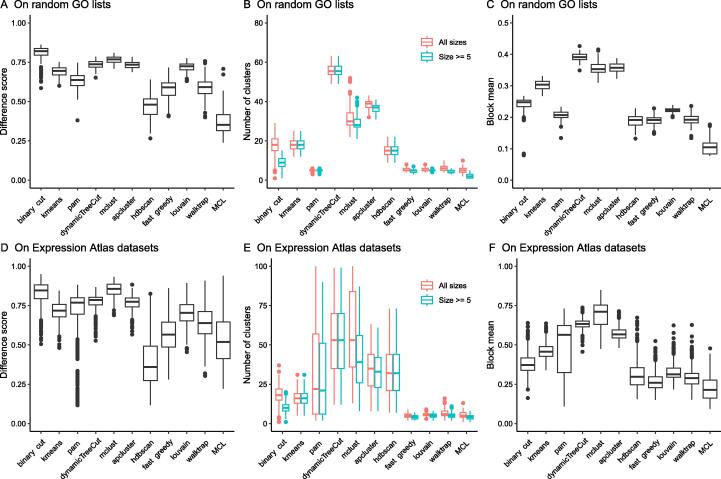
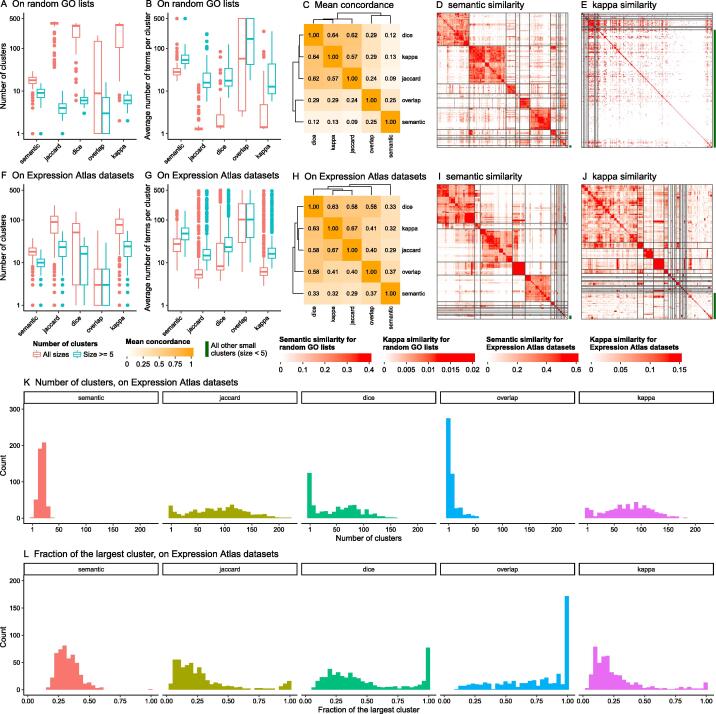
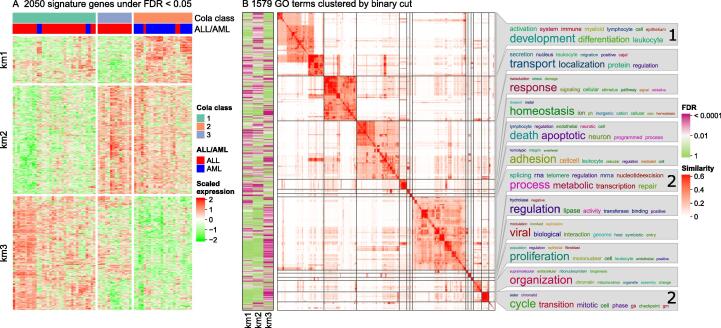
Functional enrichment analysis or gene set enrichment analysis is a basic bioinformatics method that evaluates the biological importance of a list of genes of interest. However, it may produce a long list of significant terms with highly redundant information that is difficult to summarize. Current tools to simplify enrichment results by clustering them into groups either still produce redundancy between clusters or do not retain consistent term similarities within clusters. We propose a new method named binary cut for clustering similarity matrices of functional terms. Through comprehensive benchmarks on both simulated and real-world datasets, we demonstrated that binary cut could efficiently cluster functional terms into groups where terms showed consistent similarities within groups and were mutually exclusive between groups. We compared binary cut clustering on the similarity matrices obtained from different similarity measures and found that semantic similarity worked well with binary cut, while similarity matrices based on gene overlap showed less consistent patterns. We implemented the binary cut algorithm in the R package simplifyEnrichment, which additionally provides functionalities for visualizing, summarizing, and comparing the clustering. The simplifyEnrichment package and the documentation are available at https://bioconductor.org/packages/simplifyEnrichment/.
Keywords: Clustering; Functional enrichment; R/Bioconductor; Simplify enrichment; Software; Visualization.
Copyright © 2023 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors have declared no competing interests.
Figures
Similar articles
-
DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis.Bioinformatics. 2015 Feb 15;31(4):608-9. doi: 10.1093/bioinformatics/btu684. Epub 2014 Oct 17. Bioinformatics. 2015. PMID: 25677125
-
Gene Ontology Semantic Similarity Analysis Using GOSemSim.Methods Mol Biol. 2020;2117:207-215. doi: 10.1007/978-1-0716-0301-7_11. Methods Mol Biol. 2020. PMID: 31960380
-
GOSemSim: an R package for measuring semantic similarity among GO terms and gene products.Bioinformatics. 2010 Apr 1;26(7):976-8. doi: 10.1093/bioinformatics/btq064. Epub 2010 Feb 23. Bioinformatics. 2010. PMID: 20179076
-
HCsnip: An R Package for Semi-supervised Snipping of the Hierarchical Clustering Tree.Cancer Inform. 2015 Mar 22;14:1-19. doi: 10.4137/CIN.S22080. eCollection 2015. Cancer Inform. 2015. PMID: 25861213 Free PMC article. Review.
-
multiClust: An R-package for Identifying Biologically Relevant Clusters in Cancer Transcriptome Profiles.Cancer Inform. 2016 Jun 12;15:103-14. doi: 10.4137/CIN.S38000. eCollection 2016. Cancer Inform. 2016. PMID: 27330269 Free PMC article. Review.
Cited by
-
TDP-43 chronic deficiency leads to dysregulation of transposable elements and gene expression by affecting R-loop and 5hmC crosstalk.Cell Rep. 2024 Jan 23;43(1):113662. doi: 10.1016/j.celrep.2023.113662. Epub 2024 Jan 6. Cell Rep. 2024. PMID: 38184854 Free PMC article.
-
A carbon-nitrogen negative feedback loop underlies the repeated evolution of cnidarian-Symbiodiniaceae symbioses.Nat Commun. 2023 Nov 1;14(1):6949. doi: 10.1038/s41467-023-42582-y. Nat Commun. 2023. PMID: 37914686 Free PMC article.
-
Network Pharmacology Combined with Experimental Validation Reveals the Anti-tumor Effect of Duchesnea indica against Hepatocellular Carcinoma.J Cancer. 2023 Feb 13;14(4):505-518. doi: 10.7150/jca.76591. eCollection 2023. J Cancer. 2023. PMID: 37057280 Free PMC article.
-
Characterization of Single-Cell Cis-regulatory Elements Informs Implications for Cell Differentiation.Genome Biol Evol. 2024 Nov 1;16(11):evae241. doi: 10.1093/gbe/evae241. Genome Biol Evol. 2024. PMID: 39506564 Free PMC article.
-
Identification and validation of M2 macrophage-related genes in endometriosis.Heliyon. 2023 Nov 11;9(11):e22258. doi: 10.1016/j.heliyon.2023.e22258. eCollection 2023 Nov. Heliyon. 2023. PMID: 38058639 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
