

Segmental duplications and their variation in a complete human genome
- PMID: 35357917
- PMCID: PMC8979283
- DOI: 10.1126/science.abj6965
Segmental duplications and their variation in a complete human genome
Abstract
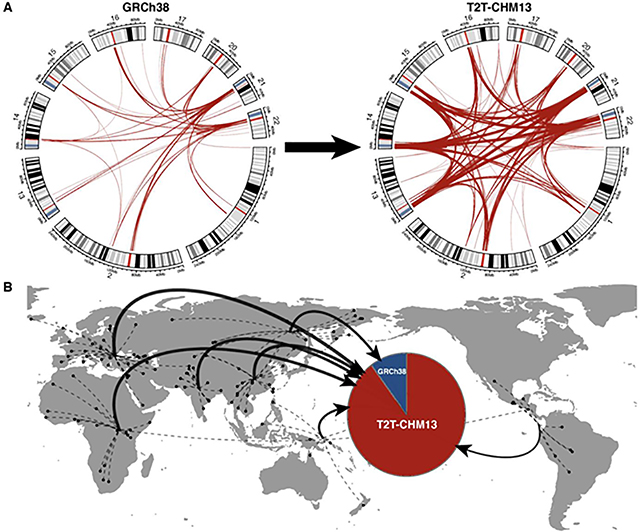
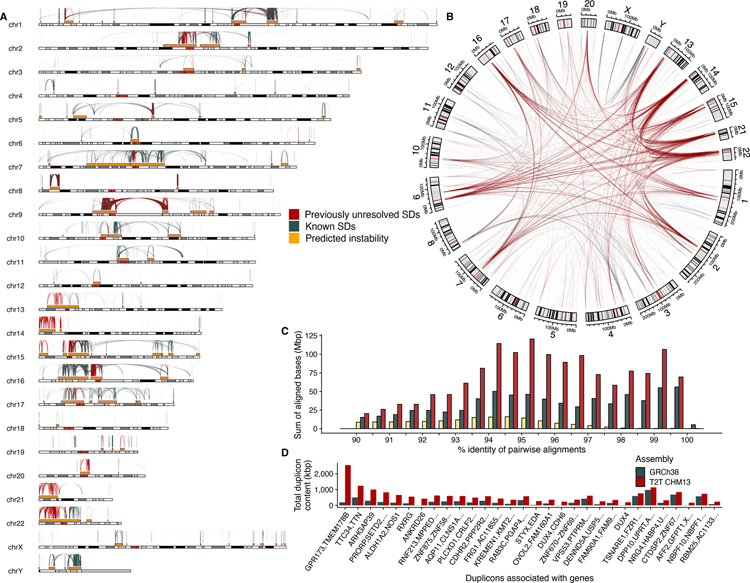
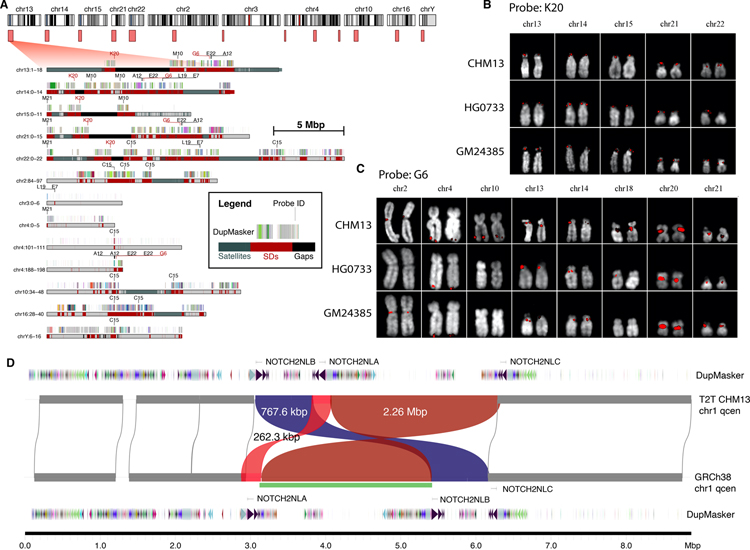
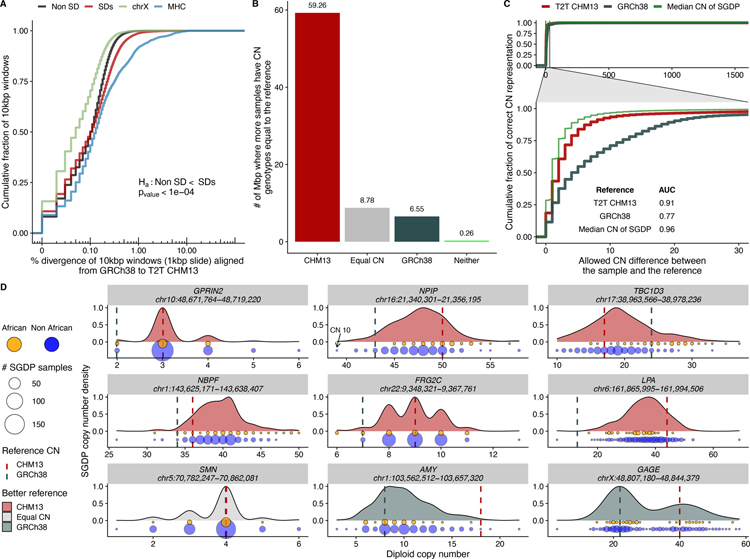
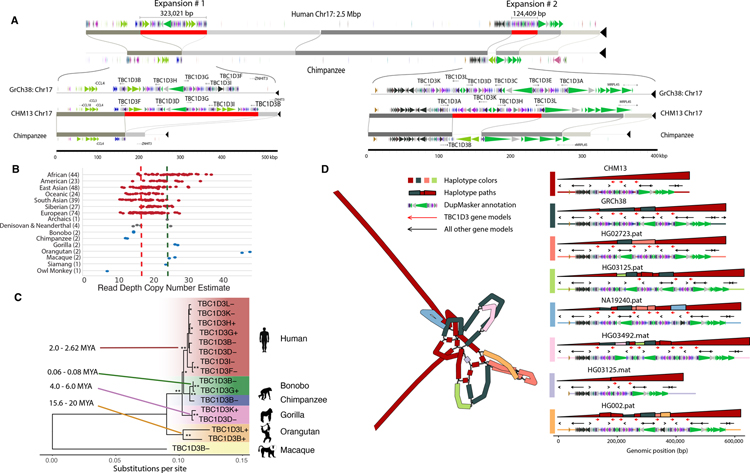
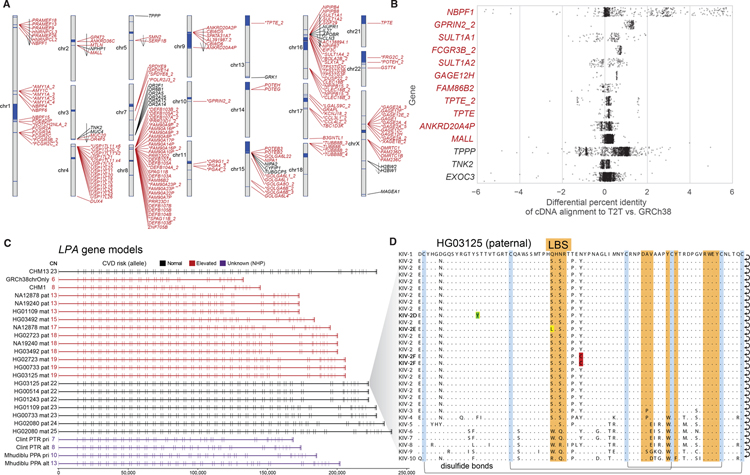
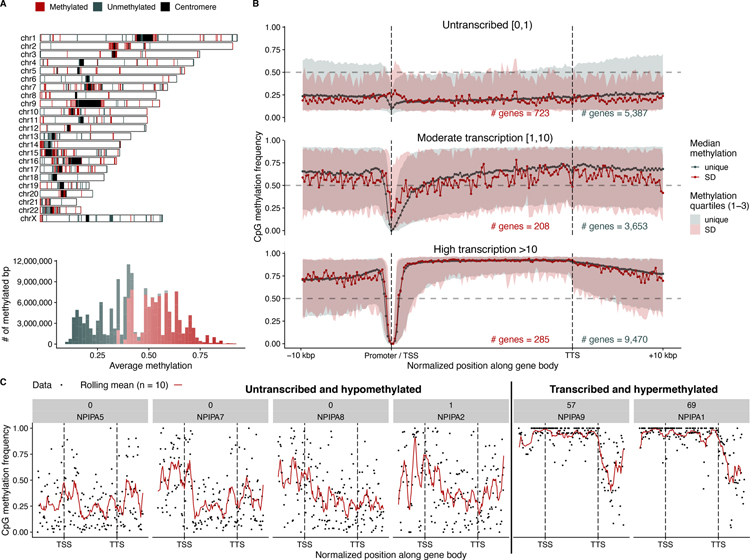
Despite their importance in disease and evolution, highly identical segmental duplications (SDs) are among the last regions of the human reference genome (GRCh38) to be fully sequenced. Using a complete telomere-to-telomere human genome (T2T-CHM13), we present a comprehensive view of human SD organization. SDs account for nearly one-third of the additional sequence, increasing the genome-wide estimate from 5.4 to 7.0% [218 million base pairs (Mbp)]. An analysis of 268 human genomes shows that 91% of the previously unresolved T2T-CHM13 SD sequence (68.3 Mbp) better represents human copy number variation. Comparing long-read assemblies from human (n = 12) and nonhuman primate (n = 5) genomes, we systematically reconstruct the evolution and structural haplotype diversity of biomedically relevant and duplicated genes. This analysis reveals patterns of structural heterozygosity and evolutionary differences in SD organization between humans and other primates.
Figures
Comment in
-
The final pieces of the human genome.Nat Rev Genet. 2022 Jun;23(6):321. doi: 10.1038/s41576-022-00494-5. Nat Rev Genet. 2022. PMID: 35488041 No abstract available.
Similar articles
-
Characterization of large-scale genomic differences in the first complete human genome.Genome Biol. 2023 Jul 4;24(1):157. doi: 10.1186/s13059-023-02995-w. Genome Biol. 2023. PMID: 37403156 Free PMC article.
-
Genome-wide maps of highly-similar intrachromosomal repeats that mediate ectopic recombination in three human genome assemblies.bioRxiv [Preprint]. 2024 Jan 31:2024.01.29.577884. doi: 10.1101/2024.01.29.577884. bioRxiv. 2024. Update in: HGG Adv. 2024 Dec 24:100396. doi: 10.1016/j.xhgg.2024.100396 PMID: 38352399 Free PMC article. Updated. Preprint.
-
Association of microsatellite pairs with segmental duplications in insect genomes.BMC Genomics. 2013 Dec 21;14:907. doi: 10.1186/1471-2164-14-907. BMC Genomics. 2013. PMID: 24359442 Free PMC article.
-
Structural variation in humans and our primate kin in the era of telomere-to-telomere genomes and pangenomics.Curr Opin Genet Dev. 2024 Aug;87:102233. doi: 10.1016/j.gde.2024.102233. Epub 2024 Jul 23. Curr Opin Genet Dev. 2024. PMID: 39042999 Free PMC article. Review.
-
The evolution of human segmental duplications and the core duplicon hypothesis.Cold Spring Harb Symp Quant Biol. 2009;74:355-62. doi: 10.1101/sqb.2009.74.011. Epub 2009 Aug 28. Cold Spring Harb Symp Quant Biol. 2009. PMID: 19717539 Free PMC article. Review.
Cited by
-
Benchmarking challenging small variants with linked and long reads.Cell Genom. 2022 May;2(5):100128. doi: 10.1016/j.xgen.2022.100128. Cell Genom. 2022. PMID: 36452119 Free PMC article.
-
Long-read sequencing and genome assembly of natural history collection samples and challenging specimens.bioRxiv [Preprint]. 2024 Sep 27:2024.03.04.583385. doi: 10.1101/2024.03.04.583385. bioRxiv. 2024. PMID: 39386456 Free PMC article. Preprint.
-
A cell type-aware framework for nominating non-coding variants in Mendelian regulatory disorders.medRxiv [Preprint]. 2023 Dec 27:2023.12.22.23300468. doi: 10.1101/2023.12.22.23300468. medRxiv. 2023. Update in: Nat Commun. 2024 Sep 27;15(1):8268. doi: 10.1038/s41467-024-52463-7 PMID: 38234731 Free PMC article. Updated. Preprint.
-
A survey of algorithms for the detection of genomic structural variants from long-read sequencing data.Nat Methods. 2023 Aug;20(8):1143-1158. doi: 10.1038/s41592-023-01932-w. Epub 2023 Jun 29. Nat Methods. 2023. PMID: 37386186 Free PMC article. Review.
-
A multilocus approach for accurate variant calling in low-copy repeats using whole-genome sequencing.Bioinformatics. 2023 Jun 30;39(39 Suppl 1):i279-i287. doi: 10.1093/bioinformatics/btad268. Bioinformatics. 2023. PMID: 37387146 Free PMC article.
References
-
- Ohno Wolf, Atkin, Evolution from fish to mammals by gene duplication. Hereditas. 59, 169–187 (1968). - PubMed
-
- Ohno, Evolution by Gene Duplication (Springer Science & Business Media, 1970).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
