

Putting Humpty Dumpty Back Together Again: What Does Protein Quantification Mean in Bottom-Up Proteomics?
- PMID: 35220718
- PMCID: PMC8976764
- DOI: 10.1021/acs.jproteome.1c00894
Putting Humpty Dumpty Back Together Again: What Does Protein Quantification Mean in Bottom-Up Proteomics?
Abstract
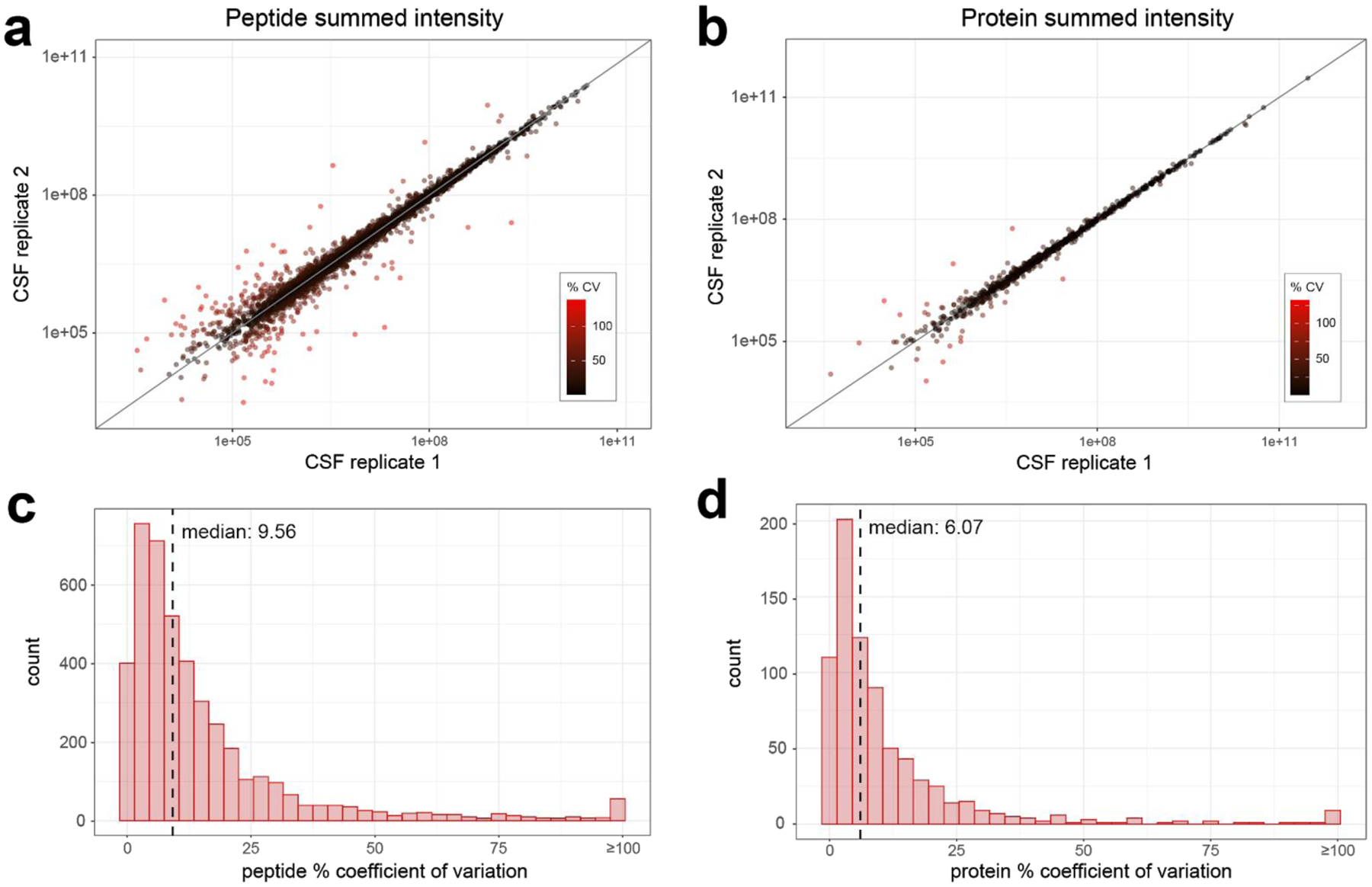
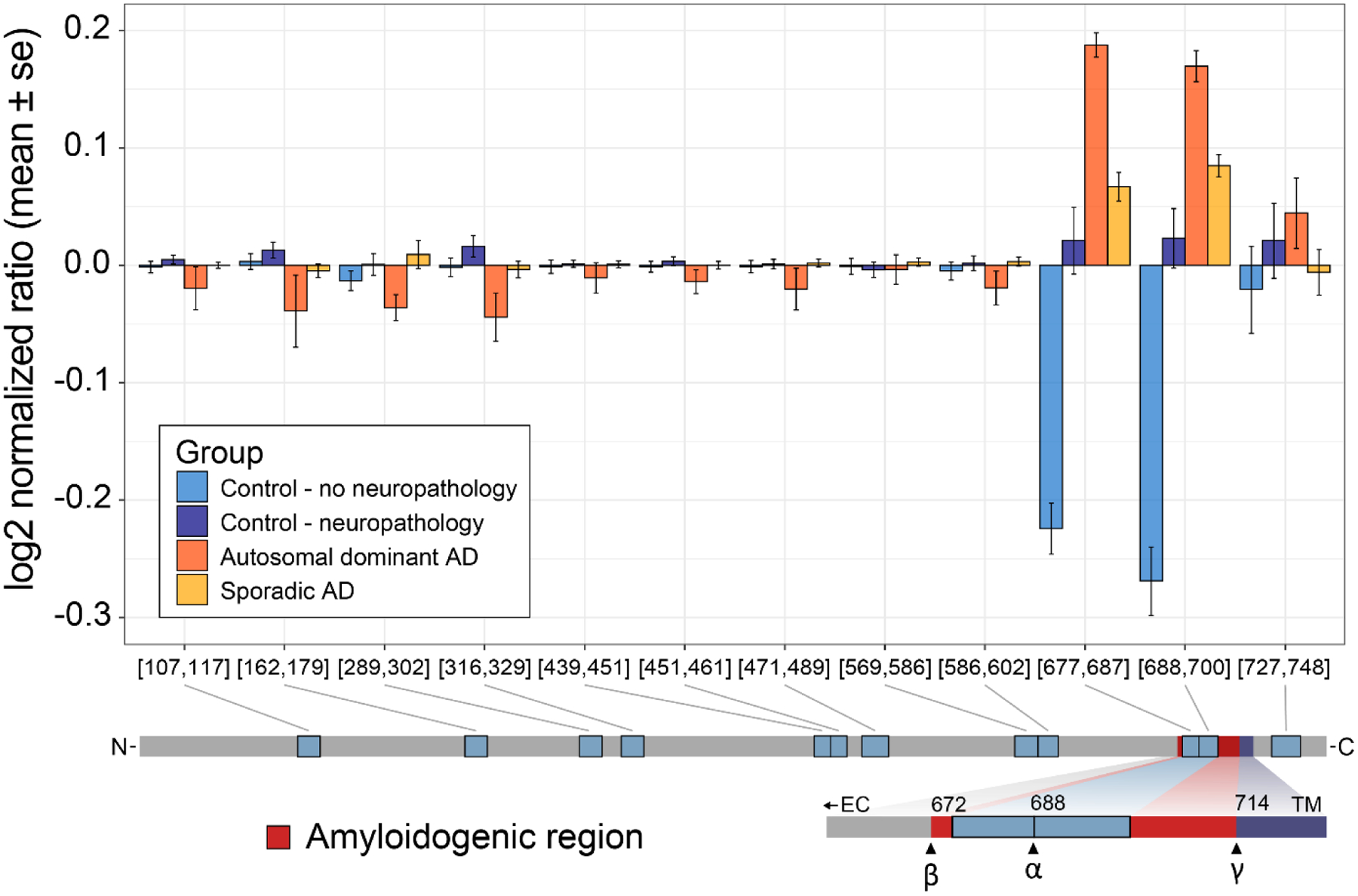
Bottom-up proteomics provides peptide measurements and has been invaluable for moving proteomics into large-scale analyses. Commonly, a single quantitative value is reported for each protein-coding gene by aggregating peptide quantities into protein groups following protein inference or parsimony. However, given the complexity of both RNA splicing and post-translational protein modification, it is overly simplistic to assume that all peptides that map to a singular protein-coding gene will demonstrate the same quantitative response. By assuming that all peptides from a protein-coding sequence are representative of the same protein, we may miss the discovery of important biological differences. To capture the contributions of existing proteoforms, we need to reconsider the practice of aggregating protein values to a single quantity per protein-coding gene.
Keywords: post-translational modifications; protein grouping; proteoforms; quantitative analysis; quantitative proteomics.
Figures



Similar articles
-
How can platelet proteomics best be used to interrogate disease?Platelets. 2023 Dec;34(1):2220046. doi: 10.1080/09537104.2023.2220046. Platelets. 2023. PMID: 37272536
-
Profiling proteoforms: promising follow-up of proteomics for biomarker discovery.Expert Rev Proteomics. 2014 Feb;11(1):121-9. doi: 10.1586/14789450.2014.878652. Expert Rev Proteomics. 2014. PMID: 24437377 Review.
-
Improving Proteoform Identifications in Complex Systems Through Integration of Bottom-Up and Top-Down Data.J Proteome Res. 2020 Aug 7;19(8):3510-3517. doi: 10.1021/acs.jproteome.0c00332. Epub 2020 Jul 10. J Proteome Res. 2020. PMID: 32584579 Free PMC article.
-
Bayesian proteoform modeling improves protein quantification of global proteomic measurements.Mol Cell Proteomics. 2014 Dec;13(12):3639-46. doi: 10.1074/mcp.M113.030932. Mol Cell Proteomics. 2014. PMID: 25433089 Free PMC article.
-
Application of targeted mass spectrometry in bottom-up proteomics for systems biology research.J Proteomics. 2018 Oct 30;189:75-90. doi: 10.1016/j.jprot.2018.02.008. Epub 2018 Feb 13. J Proteomics. 2018. PMID: 29452276 Free PMC article. Review.
Cited by
-
Imputation of cancer proteomics data with a deep model that learns from many datasets.bioRxiv [Preprint]. 2024 Aug 28:2024.08.26.609780. doi: 10.1101/2024.08.26.609780. bioRxiv. 2024. PMID: 39253518 Free PMC article. Preprint.
-
A practical introduction to holo-omics.Cell Rep Methods. 2024 Jul 15;4(7):100820. doi: 10.1016/j.crmeth.2024.100820. Epub 2024 Jul 9. Cell Rep Methods. 2024. PMID: 38986611 Free PMC article. Review.
-
Development of highly multiplex targeted proteomics assays in biofluids using the Stellar mass spectrometer.bioRxiv [Preprint]. 2024 Jun 11:2024.06.04.597431. doi: 10.1101/2024.06.04.597431. bioRxiv. 2024. PMID: 38895256 Free PMC article. Preprint.
-
Data Independent Acquisition to Inform the Development of Targeted Proteomics Assays Using a Triple Quadrupole Mass Spectrometer.bioRxiv [Preprint]. 2024 May 30:2024.05.29.596554. doi: 10.1101/2024.05.29.596554. bioRxiv. 2024. PMID: 38853953 Free PMC article. Preprint.
-
Proteomics-The State of the Field: The Definition and Analysis of Proteomes Should Be Based in Reality, Not Convenience.Proteomes. 2024 Apr 19;12(2):14. doi: 10.3390/proteomes12020014. Proteomes. 2024. PMID: 38651373 Free PMC article.
References
-
- Nesvizhskii AI, Keller A, Kolker E & Aebersold R A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem 75, 4646–4658 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
