

Haploinsufficiency of PSMD12 Causes Proteasome Dysfunction and Subclinical Autoinflammation
- PMID: 35080150
- PMCID: PMC9321778
- DOI: 10.1002/art.42070
Haploinsufficiency of PSMD12 Causes Proteasome Dysfunction and Subclinical Autoinflammation
Erratum in
-
Erratum.Arthritis Rheumatol. 2023 Jul;75(7):1138. doi: 10.1002/art.42563. Arthritis Rheumatol. 2023. PMID: 37377006 Free PMC article. No abstract available.
Abstract
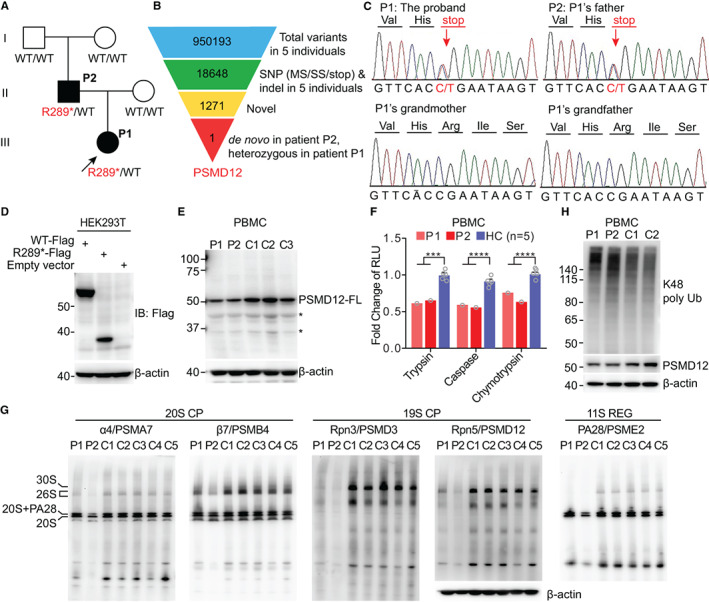
Objective: Proteasome-associated autoinflammatory syndrome (PRAAS) is caused by mutations affecting components of the proteasome and activation of the type I interferon (IFN) pathway. This study was undertaken to investigate the pathogenic mechanisms of a newly recognized type of PRAAS caused by PSMD12 haploinsufficiency.
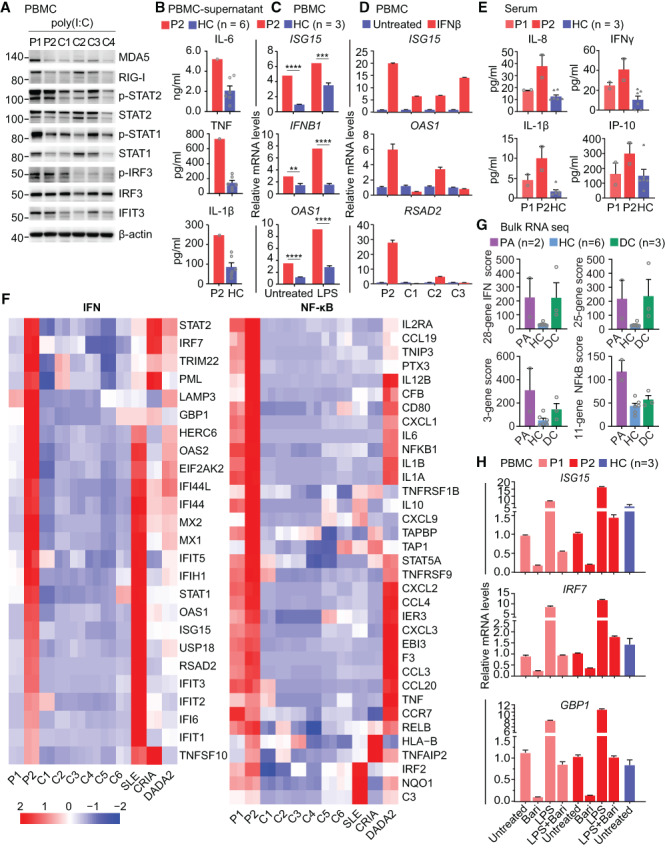
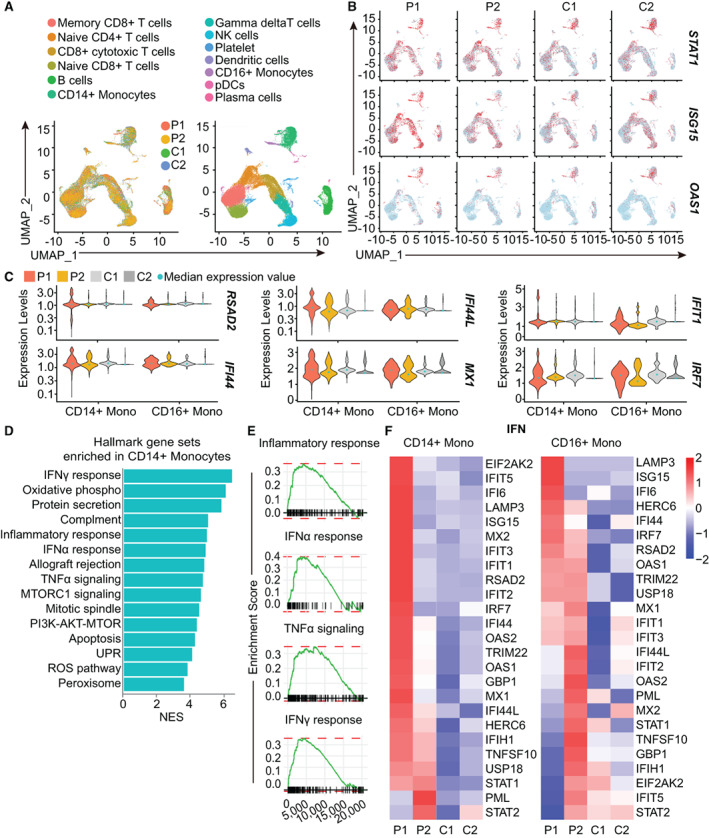
Methods: Whole-exome sequencing was performed in members of a family with skin rash, congenital uveitis, and developmental delay. We performed functional studies to assess proteasome dysfunction and inflammatory signatures in patients, and single-cell RNA sequencing to further explore the spectrum of immune cell activation.
Results: A novel truncated variant in PSMD12 (c.865C>T, p.Arg289*) was identified in 2 family members. The impairment of proteasome function was found in peripheral blood mononuclear cells (PBMCs), as well as in PSMD12-knockdown HEK 293T cell lines. Moreover, we defined the inflammatory signatures in patient PBMCs and found elevated IFN signals, especially in monocytes, by single-cell RNA sequencing.
Conclusion: These findings indicate that PSMD12 haploinsufficiency causes a set of inflammation signatures in addition to neurodevelopmental disorders. Our work expands the genotype and phenotype spectrum of PRAAS and suggests a bridge between the almost exclusively inflammatory phenotypes in the majority of PRAAS patients and the almost exclusively neurodevelopmental phenotypes in the previously reported Stankiewicz-Isidor syndrome.
© 2022 The Authors. Arthritis & Rheumatology published by Wiley Periodicals LLC on behalf of American College of Rheumatology.
Figures
Similar articles
-
PSMD12 haploinsufficiency in a neurodevelopmental disorder with autistic features.Am J Med Genet B Neuropsychiatr Genet. 2018 Dec;177(8):736-745. doi: 10.1002/ajmg.b.32688. Epub 2018 Nov 13. Am J Med Genet B Neuropsychiatr Genet. 2018. PMID: 30421579 Free PMC article.
-
Stankiewicz-Isidor syndrome: expanding the clinical and molecular phenotype.Genet Med. 2022 Jan;24(1):179-191. doi: 10.1016/j.gim.2021.09.005. Epub 2021 Nov 30. Genet Med. 2022. PMID: 34906456
-
Hematopoietic stem cell transplantation in a patient with proteasome-associated autoinflammatory syndrome (PRAAS).J Allergy Clin Immunol. 2022 Mar;149(3):1120-1127.e8. doi: 10.1016/j.jaci.2021.07.039. Epub 2021 Aug 17. J Allergy Clin Immunol. 2022. PMID: 34416217
-
Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS).Front Immunol. 2019 Nov 26;10:2756. doi: 10.3389/fimmu.2019.02756. eCollection 2019. Front Immunol. 2019. PMID: 31827472 Free PMC article. Review.
-
Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management.Int J Dermatol. 2015 Feb;54(2):121-9. doi: 10.1111/ijd.12695. Epub 2014 Dec 18. Int J Dermatol. 2015. PMID: 25521013 Review.
Cited by
-
Age-induced alterations of granulopoiesis generate atypical neutrophils that aggravate stroke pathology.Nat Immunol. 2023 Jun;24(6):925-940. doi: 10.1038/s41590-023-01505-1. Epub 2023 May 15. Nat Immunol. 2023. PMID: 37188941
-
Modulating the RPS27A/PSMD12/NF-κB pathway to control immune response in mouse brain ischemia-reperfusion injury.Mol Med. 2024 Jul 22;30(1):106. doi: 10.1186/s10020-024-00870-3. Mol Med. 2024. PMID: 39039432 Free PMC article.
-
Proteostasis Perturbations and Their Roles in Causing Sterile Inflammation and Autoinflammatory Diseases.Cells. 2022 Apr 22;11(9):1422. doi: 10.3390/cells11091422. Cells. 2022. PMID: 35563729 Free PMC article. Review.
-
Proteasome-Associated Syndromes: Updates on Genetics, Clinical Manifestations, Pathogenesis, and Treatment.J Clin Immunol. 2024 Apr 5;44(4):88. doi: 10.1007/s10875-024-01692-y. J Clin Immunol. 2024. PMID: 38578475 Review.
-
Nucleotide metabolism, leukodystrophies, and CNS pathology.J Inherit Metab Dis. 2024 Sep;47(5):860-875. doi: 10.1002/jimd.12721. Epub 2024 Feb 29. J Inherit Metab Dis. 2024. PMID: 38421058 Review.
References
-
- Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 2017;18:832–42. - PubMed
-
- Sarrabay G, Mechin D, Salhi A, Boursier G, Rittore C, Crow Y, et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J Allergy Clin Immunol 2020;145:1015–7. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
