

LEADD: Lamarckian evolutionary algorithm for de novo drug design
- PMID: 35033209
- PMCID: PMC8760751
- DOI: 10.1186/s13321-022-00582-y
LEADD: Lamarckian evolutionary algorithm for de novo drug design
Abstract
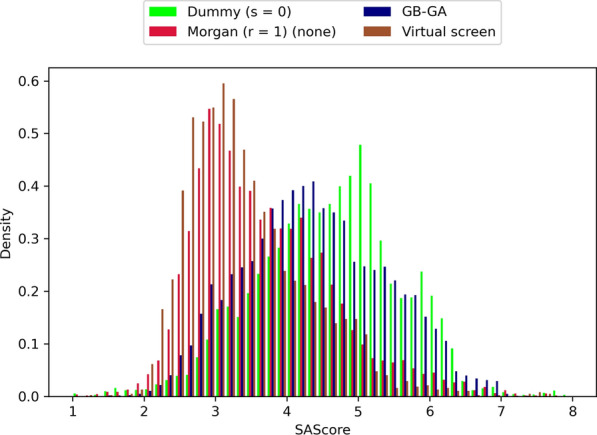
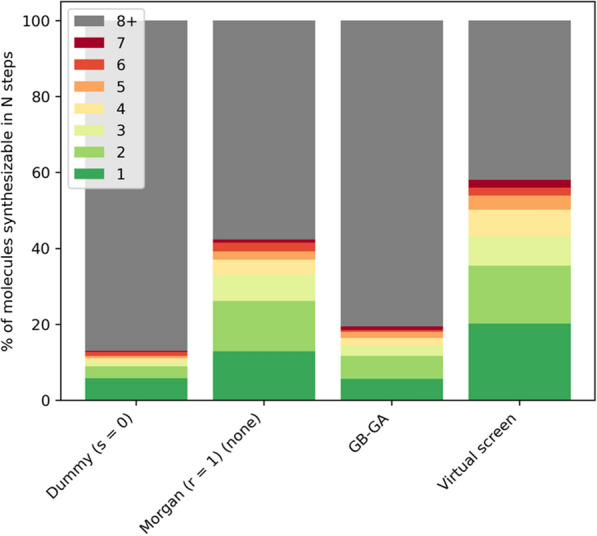
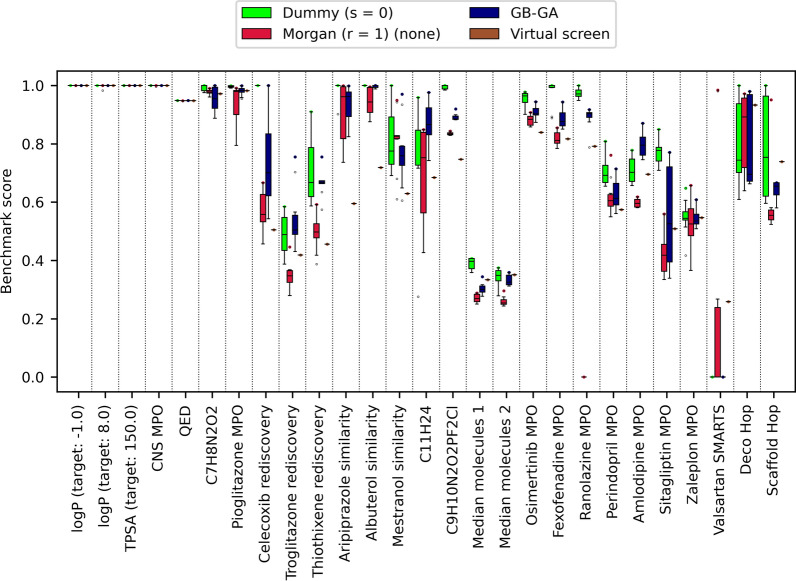
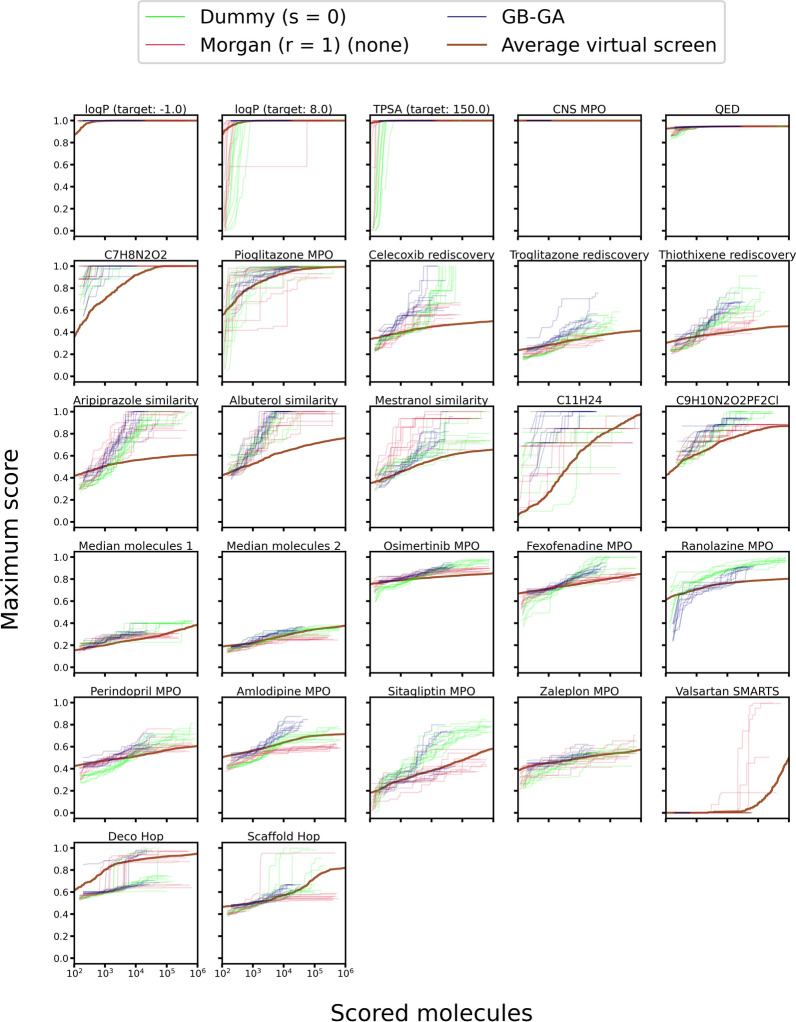
Given an objective function that predicts key properties of a molecule, goal-directed de novo molecular design is a useful tool to identify molecules that maximize or minimize said objective function. Nonetheless, a common drawback of these methods is that they tend to design synthetically unfeasible molecules. In this paper we describe a Lamarckian evolutionary algorithm for de novo drug design (LEADD). LEADD attempts to strike a balance between optimization power, synthetic accessibility of designed molecules and computational efficiency. To increase the likelihood of designing synthetically accessible molecules, LEADD represents molecules as graphs of molecular fragments, and limits the bonds that can be formed between them through knowledge-based pairwise atom type compatibility rules. A reference library of drug-like molecules is used to extract fragments, fragment preferences and compatibility rules. A novel set of genetic operators that enforce these rules in a computationally efficient manner is presented. To sample chemical space more efficiently we also explore a Lamarckian evolutionary mechanism that adapts the reproductive behavior of molecules. LEADD has been compared to both standard virtual screening and a comparable evolutionary algorithm using a standardized benchmark suite and was shown to be able to identify fitter molecules more efficiently. Moreover, the designed molecules are predicted to be easier to synthesize than those designed by other evolutionary algorithms.
Keywords: De novo drug design; Evolutionary algorithm; Fragment-based; Graph-based; Synthetic accessibility.
© 2022. The Author(s).
Conflict of interest statement
Not applicable.
Figures
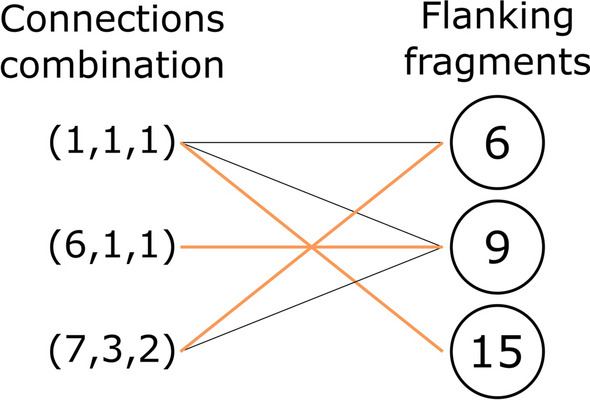

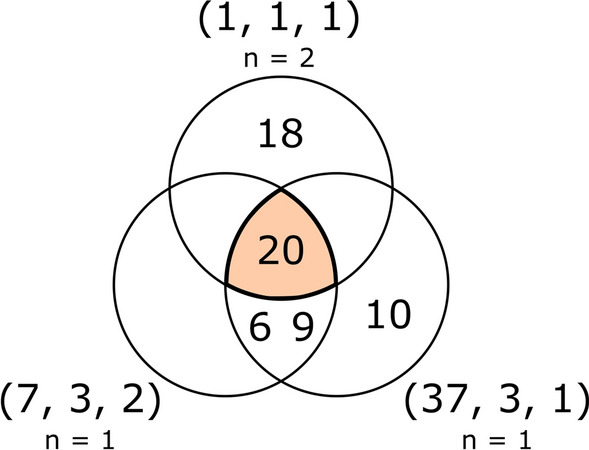
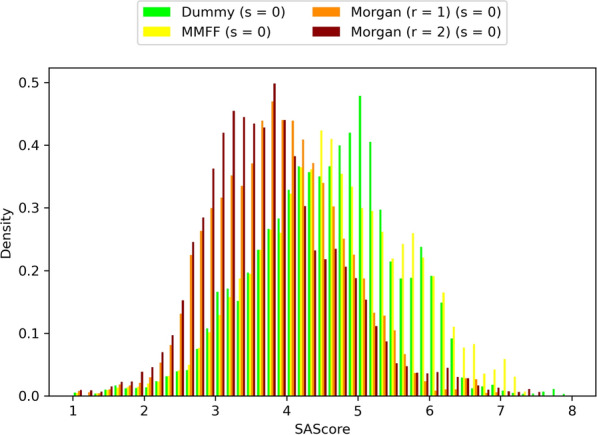
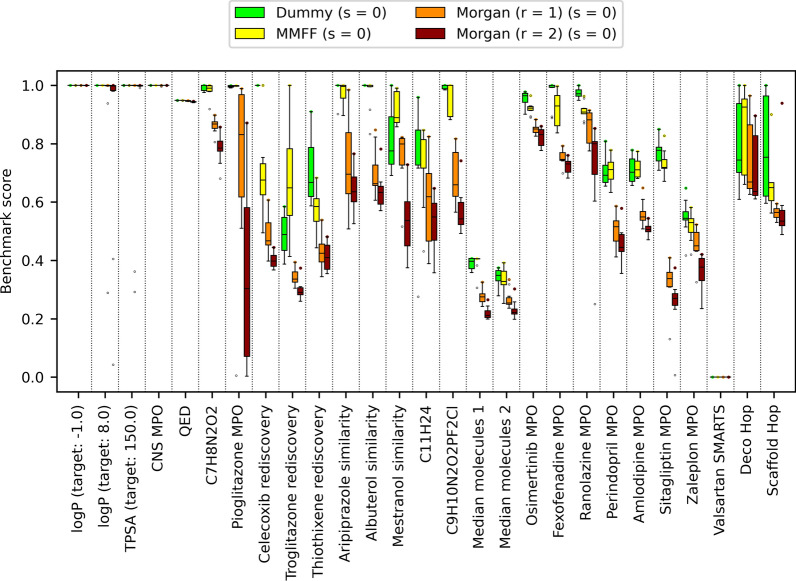
Similar articles
-
De novo drug design using multiobjective evolutionary graphs.J Chem Inf Model. 2009 Feb;49(2):295-307. doi: 10.1021/ci800308h. J Chem Inf Model. 2009. PMID: 19434831
-
RetroGNN: Fast Estimation of Synthesizability for Virtual Screening and De Novo Design by Learning from Slow Retrosynthesis Software.J Chem Inf Model. 2022 May 23;62(10):2293-2300. doi: 10.1021/acs.jcim.1c01476. Epub 2022 Apr 22. J Chem Inf Model. 2022. PMID: 35452226 Review.
-
RENATE: A Pseudo-retrosynthetic Tool for Synthetically Accessible de novo Design.Mol Inform. 2022 Apr;41(4):e2100207. doi: 10.1002/minf.202100207. Epub 2021 Nov 8. Mol Inform. 2022. PMID: 34750989 Free PMC article.
-
MoleGear: A Java-Based Platform for Evolutionary De Novo Molecular Design.Molecules. 2019 Apr 11;24(7):1444. doi: 10.3390/molecules24071444. Molecules. 2019. PMID: 30979097 Free PMC article.
-
Recent Advances in Automated Structure-Based De Novo Drug Design.J Chem Inf Model. 2024 Mar 25;64(6):1794-1805. doi: 10.1021/acs.jcim.4c00247. Epub 2024 Mar 14. J Chem Inf Model. 2024. PMID: 38485516 Free PMC article. Review.
Cited by
-
Galileo: Three-dimensional searching in large combinatorial fragment spaces on the example of pharmacophores.J Comput Aided Mol Des. 2023 Jan;37(1):1-16. doi: 10.1007/s10822-022-00485-y. Epub 2022 Nov 24. J Comput Aided Mol Des. 2023. PMID: 36418668 Free PMC article.
-
Molecule auto-correction to facilitate molecular design.J Comput Aided Mol Des. 2024 Feb 16;38(1):10. doi: 10.1007/s10822-024-00549-1. J Comput Aided Mol Des. 2024. PMID: 38363377 Free PMC article.
-
Selection of Mexican Medicinal Plants by Identification of Potential Phytochemicals with Anti-Aging, Anti-Inflammatory, and Anti-Oxidant Properties through Network Analysis and Chemoinformatic Screening.Biomolecules. 2023 Nov 20;13(11):1673. doi: 10.3390/biom13111673. Biomolecules. 2023. PMID: 38002355 Free PMC article.
-
EMBL's European Bioinformatics Institute (EMBL-EBI) in 2022.Nucleic Acids Res. 2023 Jan 6;51(D1):D9-D17. doi: 10.1093/nar/gkac1098. Nucleic Acids Res. 2023. PMID: 36477213 Free PMC article.
-
A molecule perturbation software library and its application to study the effects of molecular design constraints.J Cheminform. 2023 Sep 26;15(1):89. doi: 10.1186/s13321-023-00761-5. J Cheminform. 2023. PMID: 37752561 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
