

Subcellular proteomics
- PMID: 34549195
- PMCID: PMC8451152
- DOI: 10.1038/s43586-021-00029-y
Subcellular proteomics
Abstract
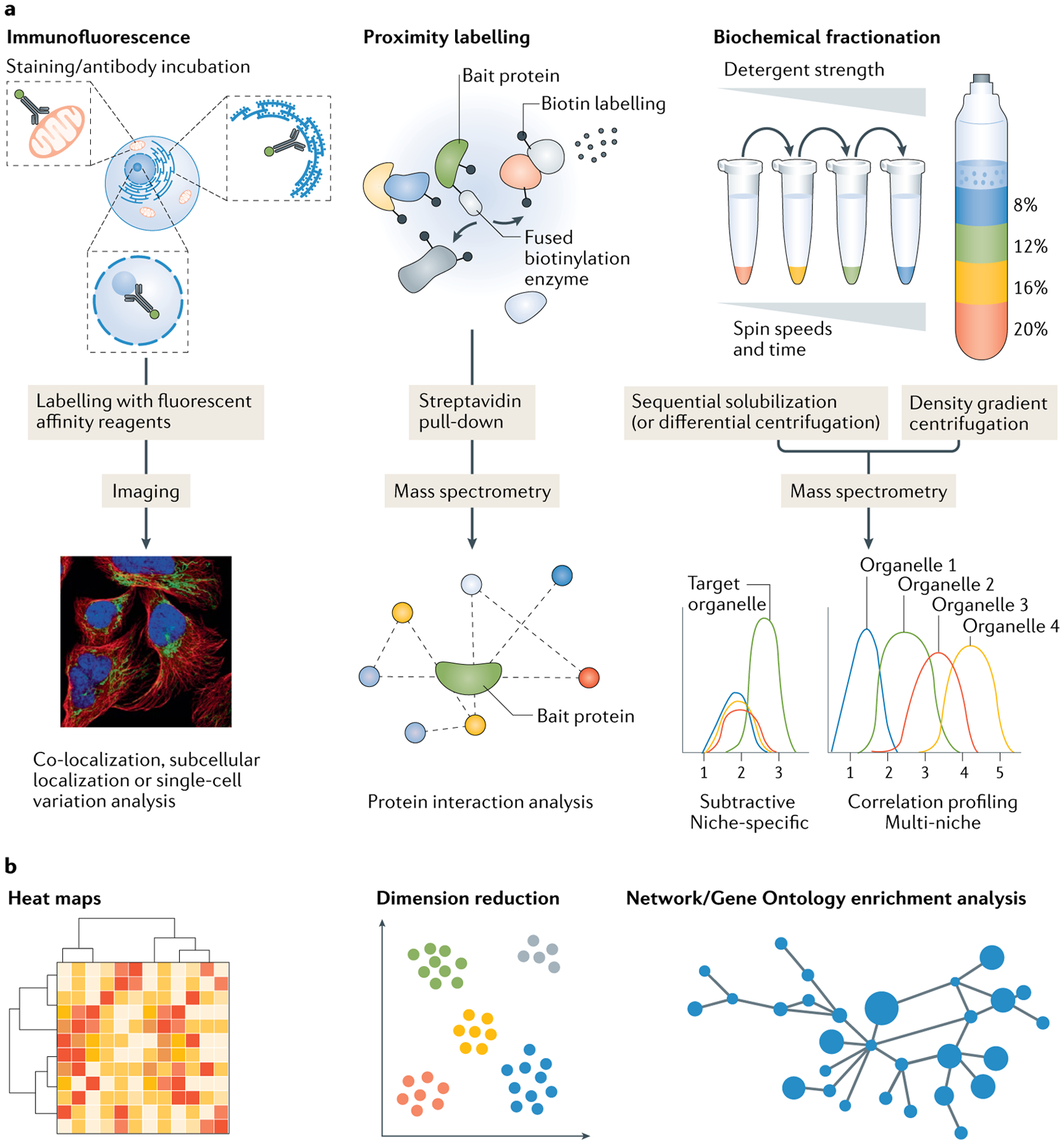
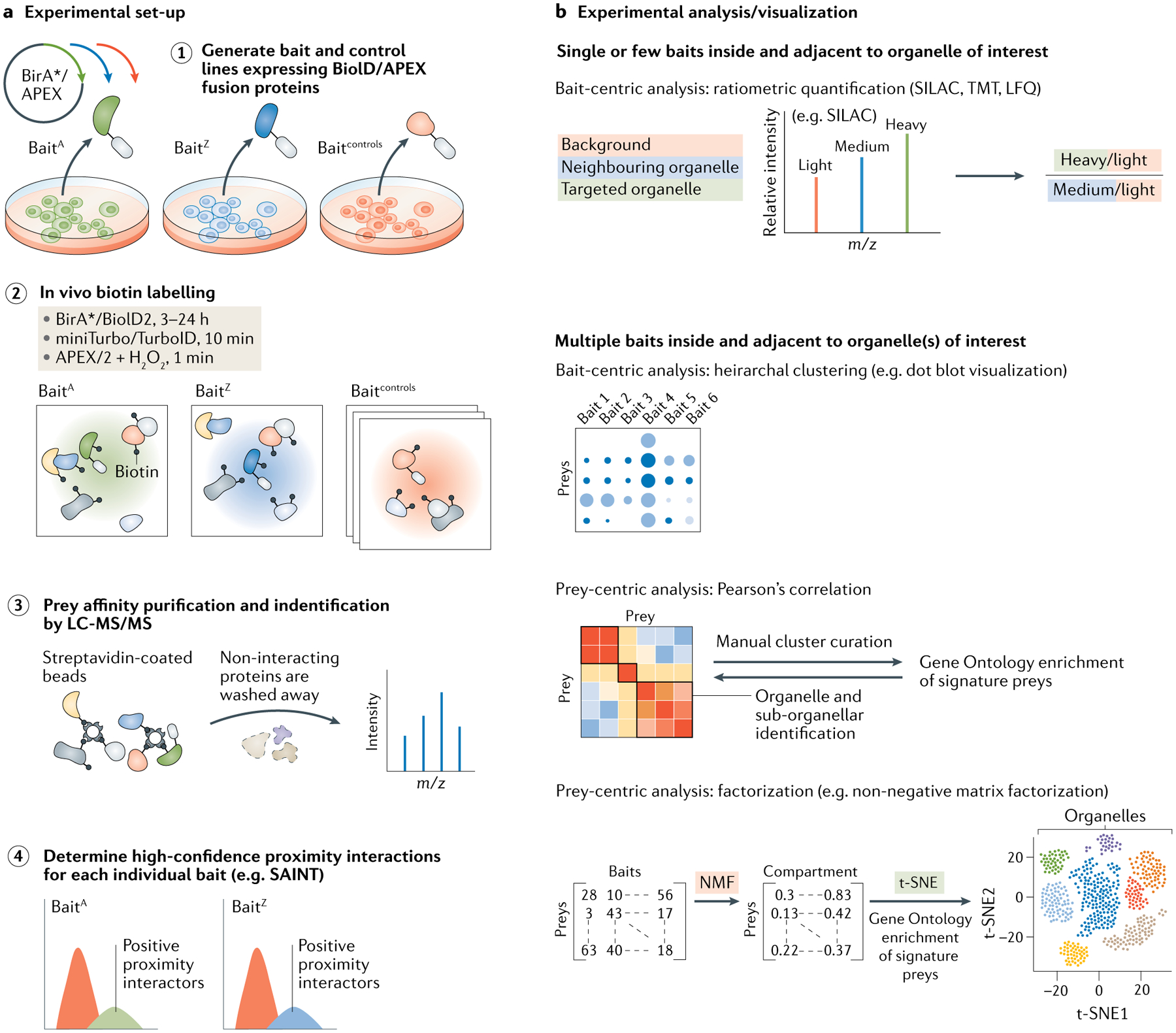
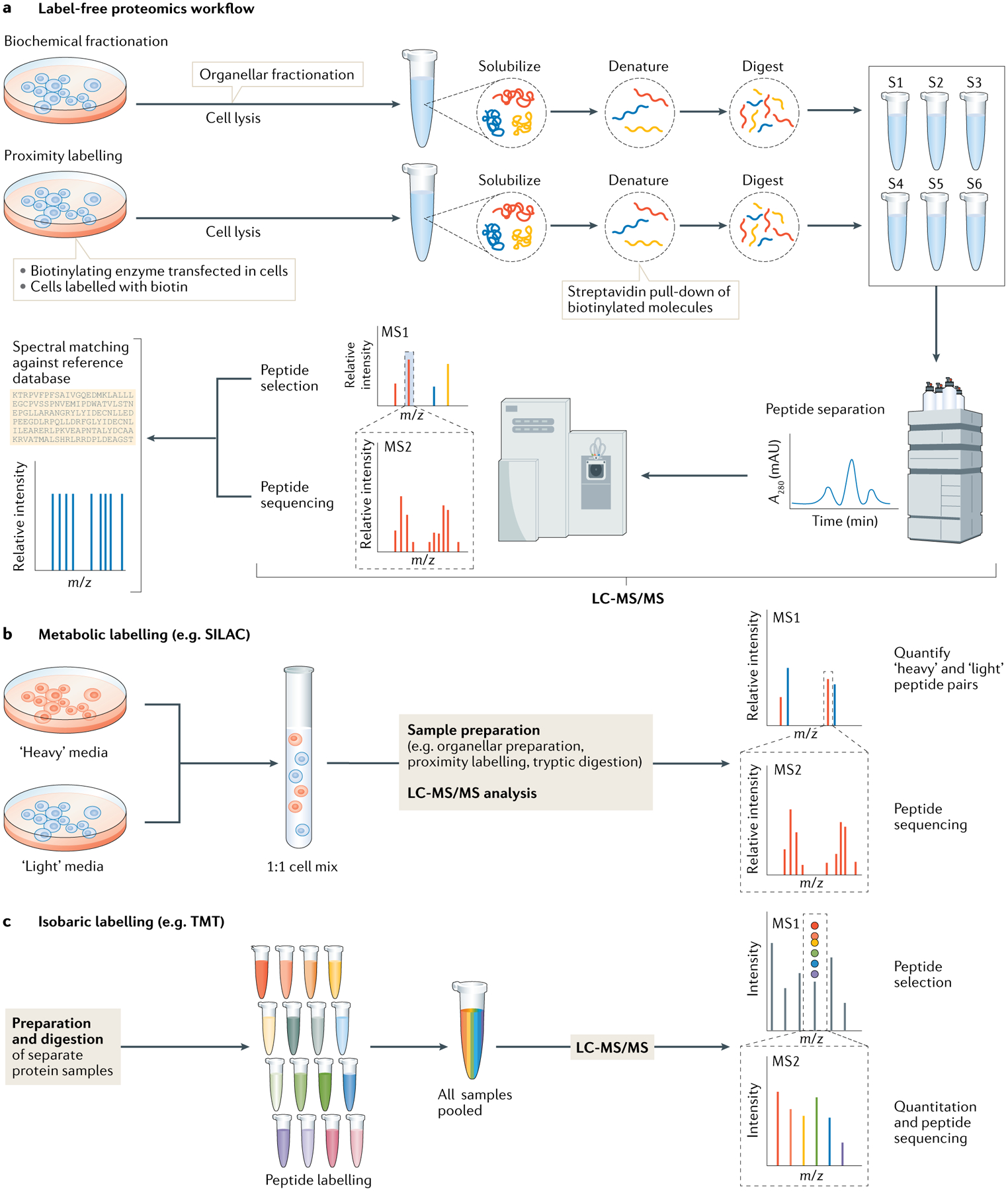
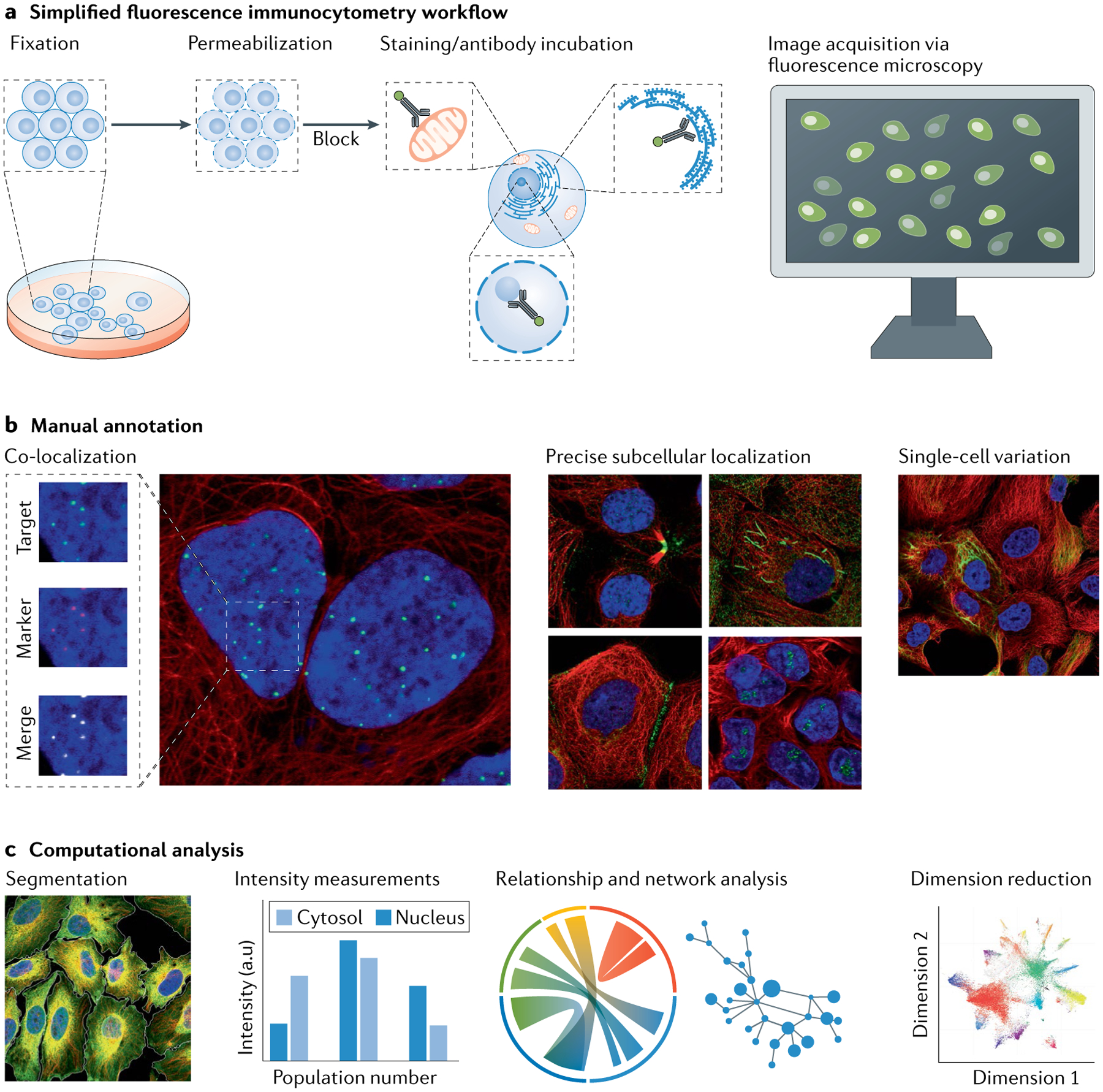
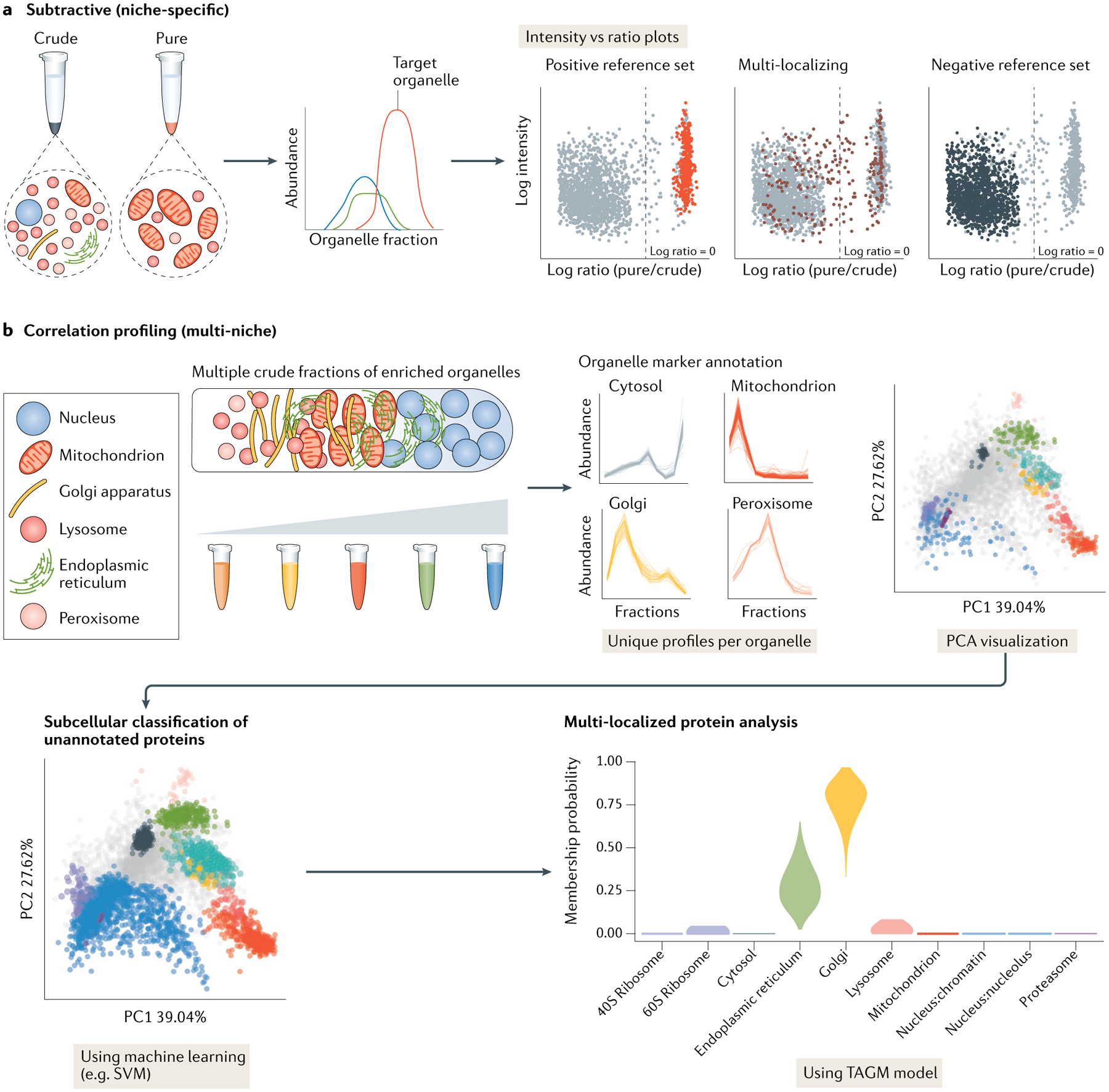
The eukaryotic cell is compartmentalized into subcellular niches, including membrane-bound and membrane-less organelles. Proteins localize to these niches to fulfil their function, enabling discreet biological processes to occur in synchrony. Dynamic movement of proteins between niches is essential for cellular processes such as signalling, growth, proliferation, motility and programmed cell death, and mutations causing aberrant protein localization are associated with a wide range of diseases. Determining the location of proteins in different cell states and cell types and how proteins relocalize following perturbation is important for understanding their functions, related cellular processes and pathologies associated with their mislocalization. In this Primer, we cover the major spatial proteomics methods for determining the location, distribution and abundance of proteins within subcellular structures. These technologies include fluorescent imaging, protein proximity labelling, organelle purification and cell-wide biochemical fractionation. We describe their workflows, data outputs and applications in exploring different cell biological scenarios, and discuss their main limitations. Finally, we describe emerging technologies and identify areas that require technological innovation to allow better characterization of the spatial proteome.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures
Similar articles
-
Recent Advancements in Subcellular Proteomics: Growing Impact of Organellar Protein Niches on the Understanding of Cell Biology.J Proteome Res. 2024 Aug 2;23(8):2700-2722. doi: 10.1021/acs.jproteome.3c00839. Epub 2024 Mar 7. J Proteome Res. 2024. PMID: 38451675 Free PMC article. Review.
-
Dynamic Organellar Maps for Spatial Proteomics.Curr Protoc Cell Biol. 2019 Jun;83(1):e81. doi: 10.1002/cpcb.81. Epub 2018 Nov 29. Curr Protoc Cell Biol. 2019. PMID: 30489039
-
Spatial proteomics of vesicular trafficking: coupling mass spectrometry and imaging approaches in membrane biology.Plant Biotechnol J. 2023 Feb;21(2):250-269. doi: 10.1111/pbi.13929. Epub 2022 Nov 4. Plant Biotechnol J. 2023. PMID: 36204821 Free PMC article. Review.
-
Advances in spatial proteomics: Mapping proteome architecture from protein complexes to subcellular localizations.Cell Chem Biol. 2024 Sep 19;31(9):1665-1687. doi: 10.1016/j.chembiol.2024.08.008. Cell Chem Biol. 2024. PMID: 39303701 Review.
-
Proteomics methods for subcellular proteome analysis.FEBS J. 2013 Nov;280(22):5626-34. doi: 10.1111/febs.12502. Epub 2013 Sep 20. FEBS J. 2013. PMID: 24034475 Review.
Cited by
-
CELL-E 2: Translating Proteins to Pictures and Back with a Bidirectional Text-to-Image Transformer.Adv Neural Inf Process Syst. 2023 Dec;36:4899-4914. Adv Neural Inf Process Syst. 2023. PMID: 39021511 Free PMC article.
-
Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of protein homeostasis disruptions.bioRxiv [Preprint]. 2024 Jan 17:2023.01.04.521821. doi: 10.1101/2023.01.04.521821. bioRxiv. 2024. Update in: Nat Commun. 2024 Mar 11;15(1):2207. doi: 10.1038/s41467-024-46600-5 PMID: 36711879 Free PMC article. Updated. Preprint.
-
Druggability of Targets for Diagnostic Radiopharmaceuticals.ACS Pharmacol Transl Sci. 2023 Jul 12;6(8):1107-1119. doi: 10.1021/acsptsci.3c00081. eCollection 2023 Aug 11. ACS Pharmacol Transl Sci. 2023. PMID: 37588760 Free PMC article. Review.
-
Is a spice missing from the recipe? The intra-cellular localization of vanillin biosynthesis needs further investigations.Plant Biol (Stuttg). 2023 Jan;25(1):3-7. doi: 10.1111/plb.13465. Epub 2022 Sep 16. Plant Biol (Stuttg). 2023. PMID: 36066305 Free PMC article.
-
Recent Advances in the Prediction of Subcellular Localization of Proteins and Related Topics.Front Bioinform. 2022 May 19;2:910531. doi: 10.3389/fbinf.2022.910531. eCollection 2022. Front Bioinform. 2022. PMID: 36304291 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources