

Targeting protein-protein interactions in the DNA damage response pathways for cancer chemotherapy
- PMID: 34458830
- PMCID: PMC8342002
- DOI: 10.1039/d1cb00101a
Targeting protein-protein interactions in the DNA damage response pathways for cancer chemotherapy
Abstract
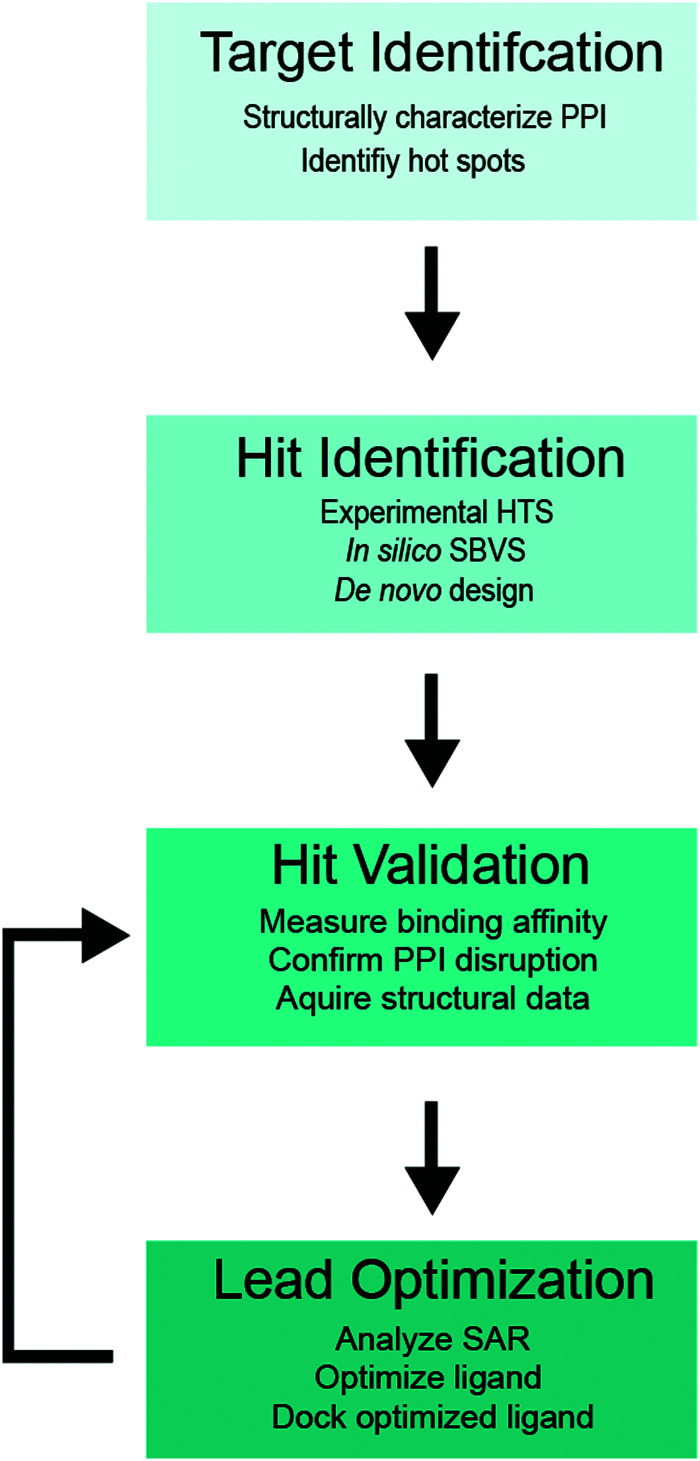
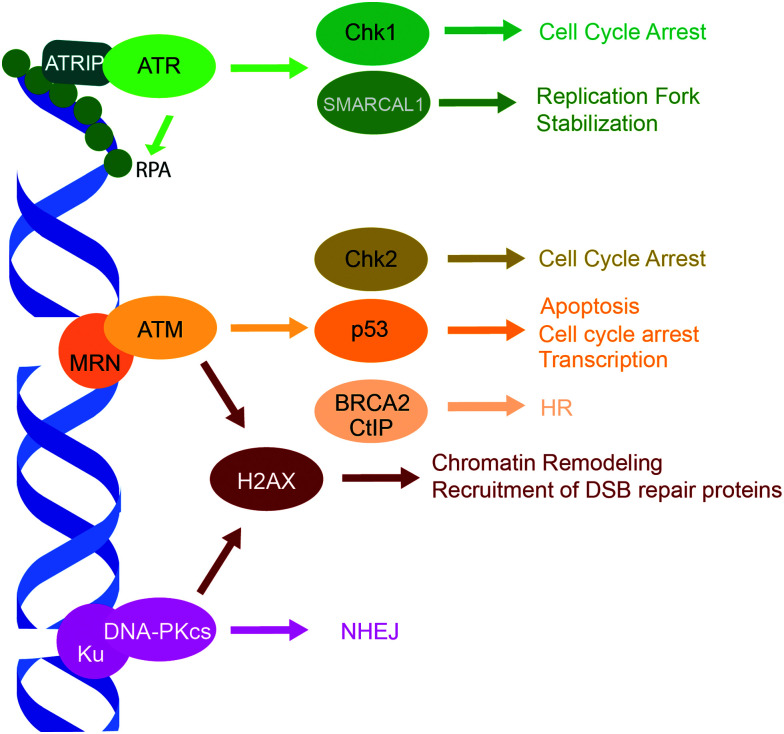
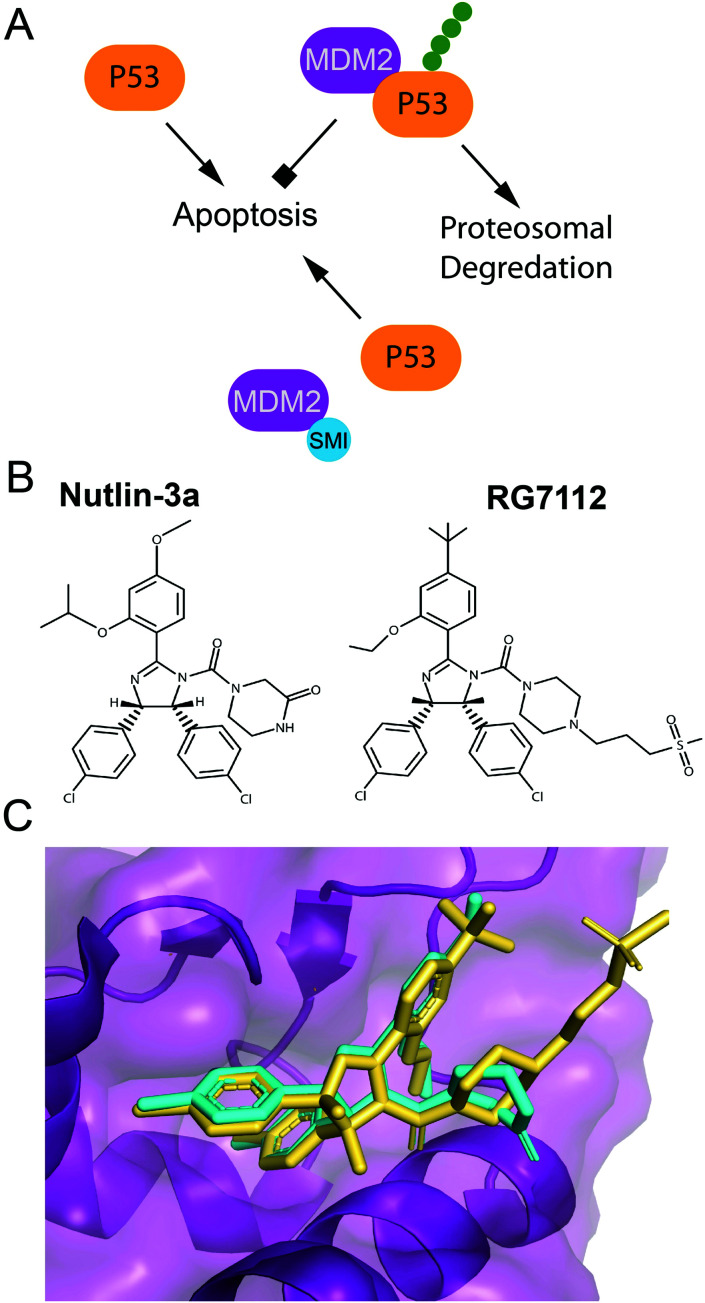
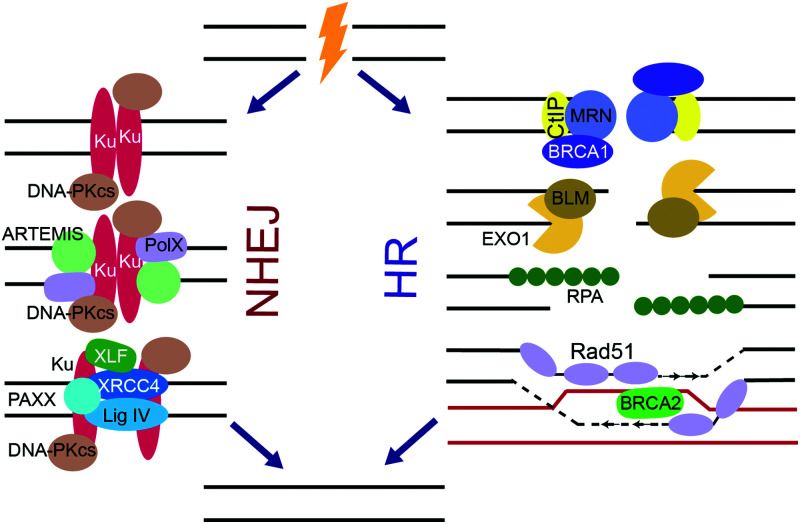
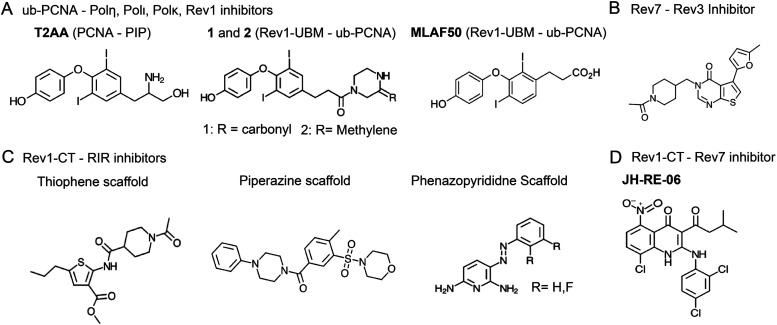
Cellular DNA damage response (DDR) is an extensive signaling network that orchestrates DNA damage recognition, repair and avoidance, cell cycle progression and cell death. DDR alteration is a hallmark of cancer, with the deficiency in one DDR capability often compensated by a dependency on alternative pathways endowing cancer cells with survival and growth advantage. Targeting these DDR pathways has provided multiple opportunities for the development of cancer therapies. Traditional drug discovery has mainly focused on catalytic inhibitors that block enzyme active sites, which limits the number of potential drug targets within the DDR pathways. This review article describes the emerging approach to the development of cancer therapeutics targeting essential protein-protein interactions (PPIs) in the DDR network. The overall strategy for the structure-based design of small molecule PPI inhibitors is discussed, followed by an overview of the major DNA damage sensing, DNA repair, and DNA damage tolerance pathways with a specific focus on PPI targets for anti-cancer drug design. The existing small molecule inhibitors of DDR PPIs are summarized that selectively kill cancer cells and/or sensitize cancers to front-line genotoxic therapies, and a range of new PPI targets are proposed that may lead to the development of novel chemotherapeutics.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures
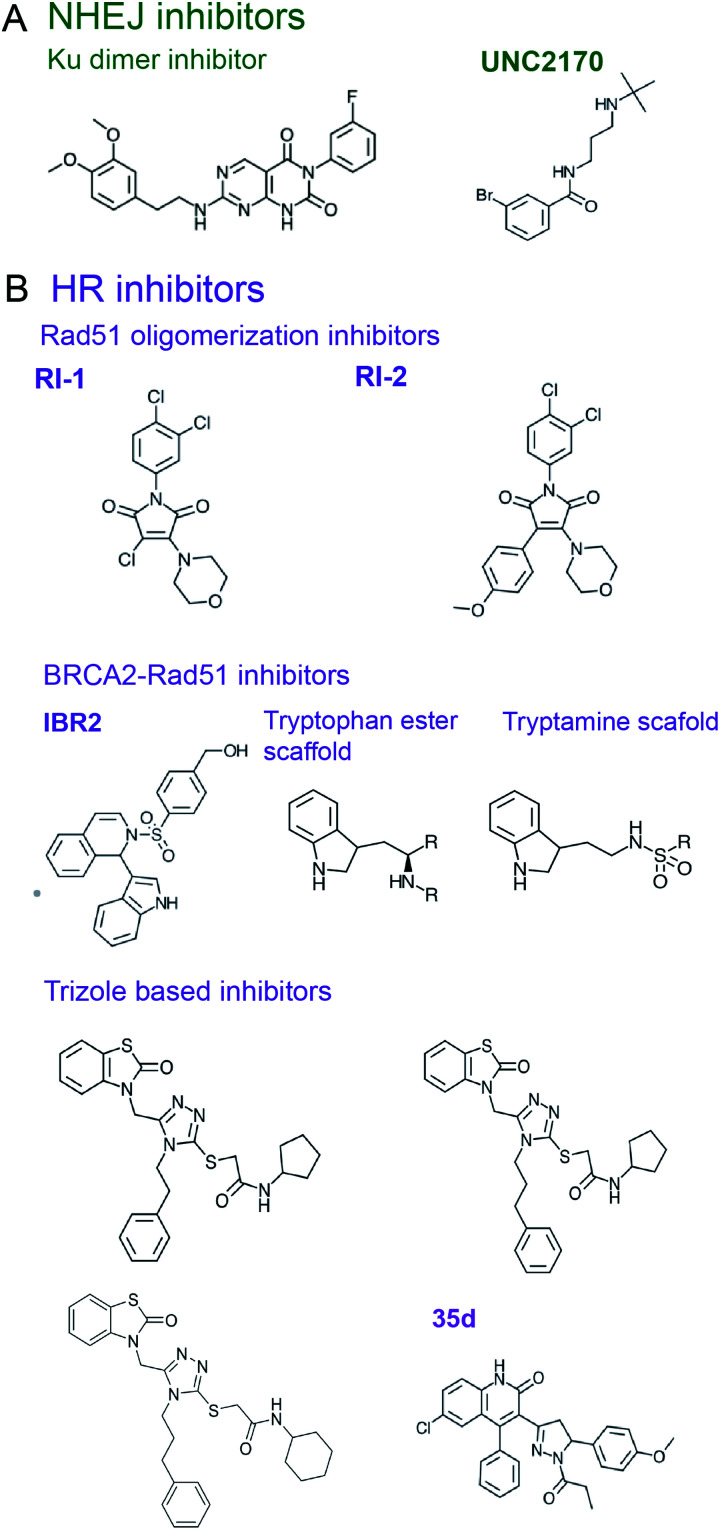
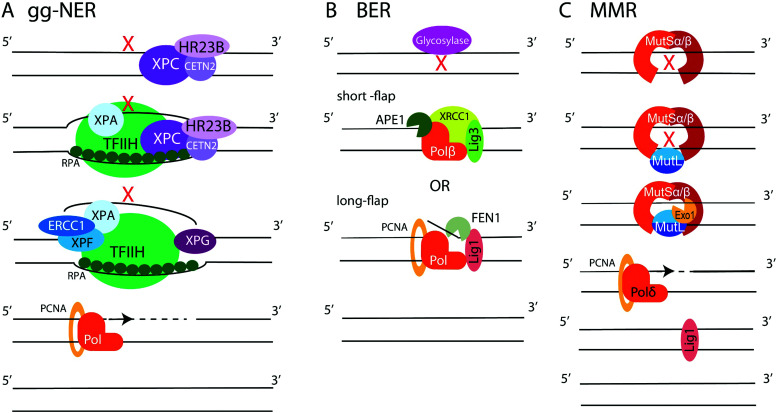
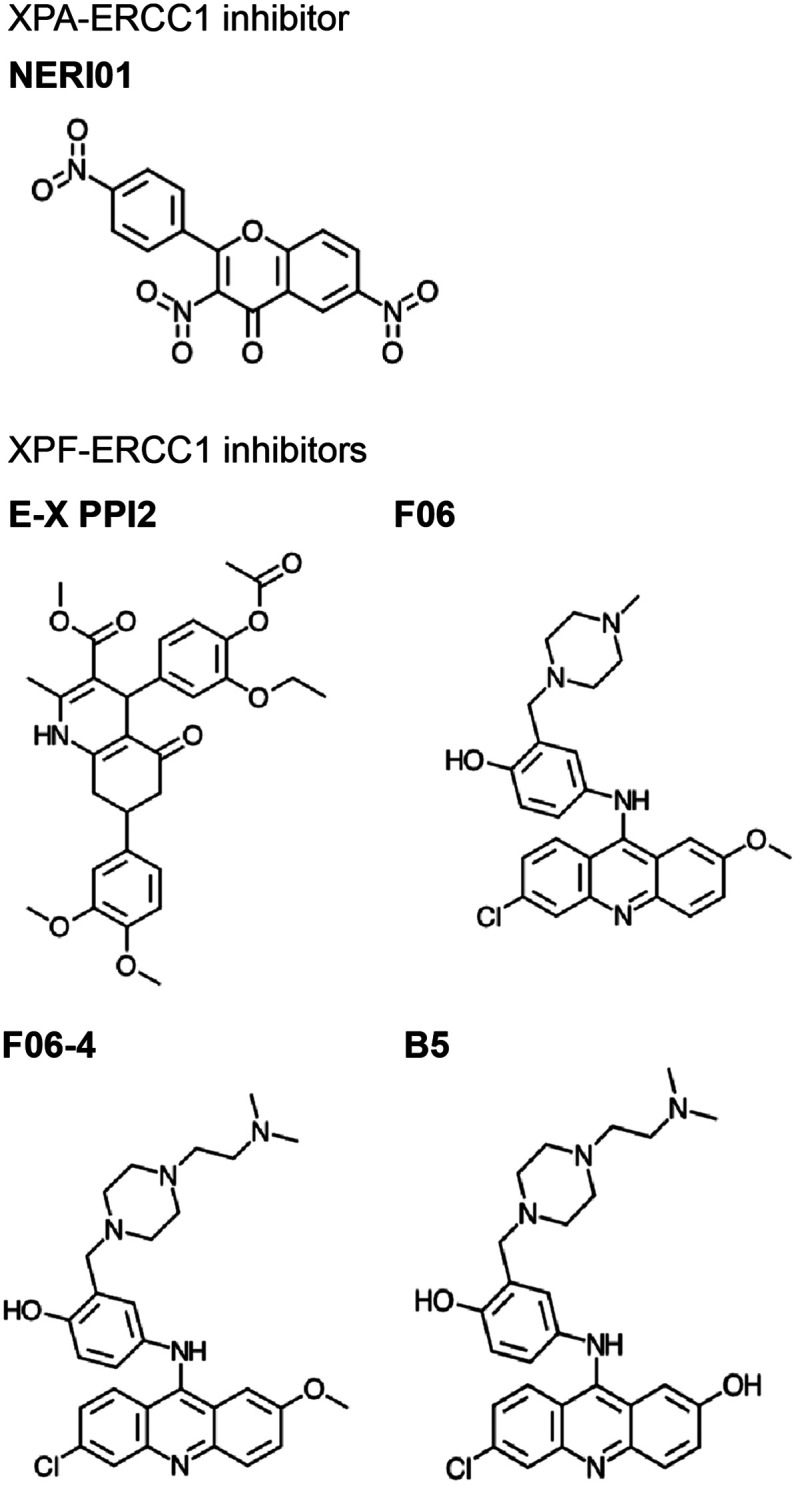
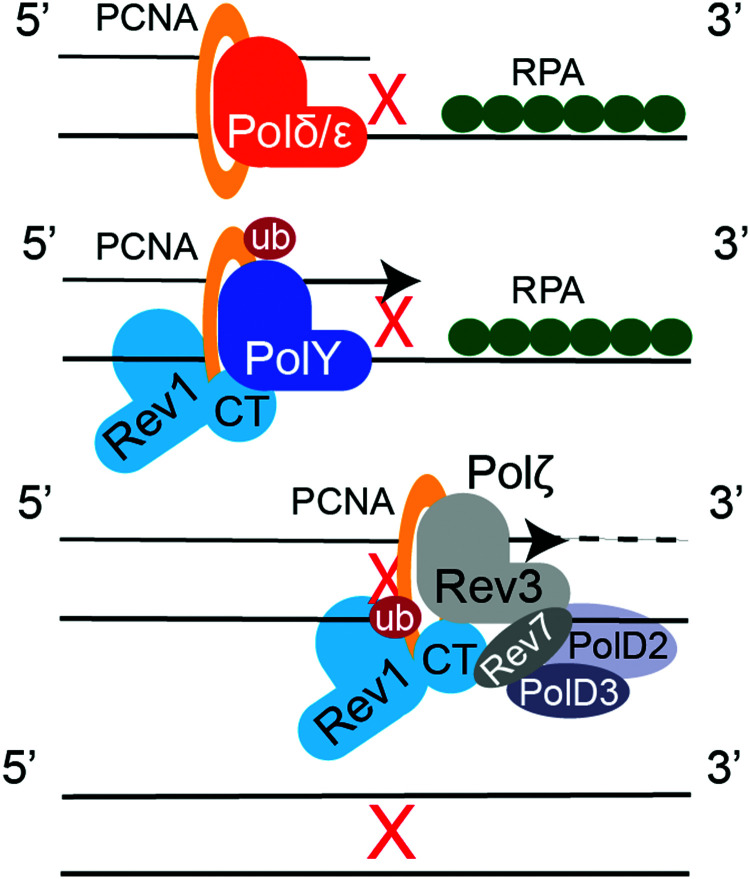


Similar articles
-
Noncoding RNAs in DNA Damage Response: Opportunities for Cancer Therapeutics.Methods Mol Biol. 2018;1699:3-21. doi: 10.1007/978-1-4939-7435-1_1. Methods Mol Biol. 2018. PMID: 29086365
-
Understanding DNA damage response and DNA repair pathways: applications to more targeted cancer therapeutics.Semin Oncol. 2009 Apr;36(2 Suppl 1):S42-51. doi: 10.1053/j.seminoncol.2009.02.004. Semin Oncol. 2009. PMID: 19393835
-
Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies.Ther Adv Med Oncol. 2018 Jul 13;10:1758835918786658. doi: 10.1177/1758835918786658. eCollection 2018. Ther Adv Med Oncol. 2018. PMID: 30023007 Free PMC article. Review.
-
Role of deubiquitinases in DNA damage response.DNA Repair (Amst). 2019 Apr;76:89-98. doi: 10.1016/j.dnarep.2019.02.011. Epub 2019 Feb 21. DNA Repair (Amst). 2019. PMID: 30831436 Free PMC article. Review.
-
Therapeutic Targeting of DNA Repair Pathways in Pediatric Extracranial Solid Tumors: Current State and Implications for Immunotherapy.Cancers (Basel). 2024 Apr 25;16(9):1648. doi: 10.3390/cancers16091648. Cancers (Basel). 2024. PMID: 38730598 Free PMC article. Review.
Cited by
-
Targeting the hSSB1-INTS3 Interface: A Computational Screening Driven Approach to Identify Potential Modulators.ACS Omega. 2024 Feb 8;9(7):8362-8373. doi: 10.1021/acsomega.3c09267. eCollection 2024 Feb 20. ACS Omega. 2024. PMID: 38405517 Free PMC article.
-
Targeting the DNA Damage Response for Cancer Therapy.Int J Mol Sci. 2023 Nov 2;24(21):15907. doi: 10.3390/ijms242115907. Int J Mol Sci. 2023. PMID: 37958890 Free PMC article. Review.
-
Photonic Crystal Surface Mode Real-Time Imaging of RAD51 DNA Repair Protein Interaction with the ssDNA Substrate.Biosensors (Basel). 2024 Jan 14;14(1):43. doi: 10.3390/bios14010043. Biosensors (Basel). 2024. PMID: 38248420 Free PMC article.
-
Identification of hub genes involved in cisplatin resistance in head and neck cancer.J Genet Eng Biotechnol. 2023 Jan 30;21(1):9. doi: 10.1186/s43141-023-00468-y. J Genet Eng Biotechnol. 2023. PMID: 36715825 Free PMC article.
-
The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome.Cells. 2021 Sep 29;10(10):2597. doi: 10.3390/cells10102597. Cells. 2021. PMID: 34685575 Free PMC article. Review.
References
-
- Friedberg E. C., Walker G. C., Siede W. and Wood R. D., DNA repair and mutagenesis, American Society for Microbiology Press, 2005
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources