

Nonrefoldability is Pervasive Across the E. coli Proteome
- PMID: 34308638
- PMCID: PMC8650709
- DOI: 10.1021/jacs.1c03270
Nonrefoldability is Pervasive Across the E. coli Proteome
Abstract
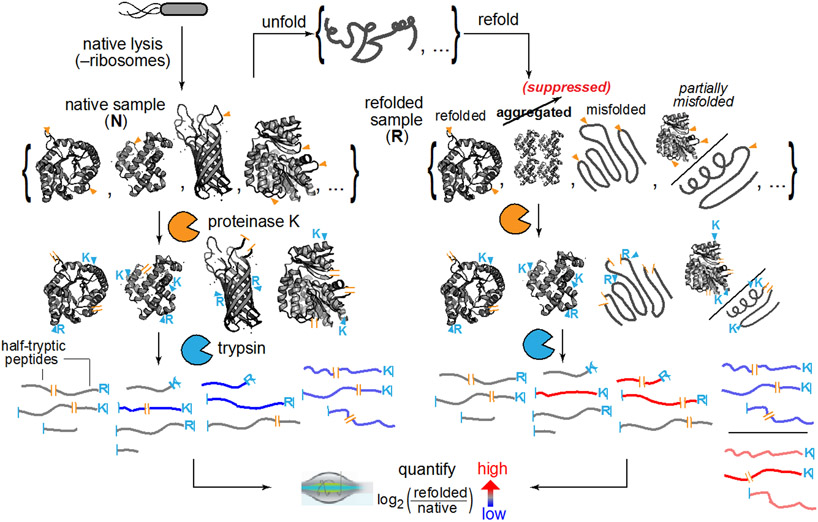
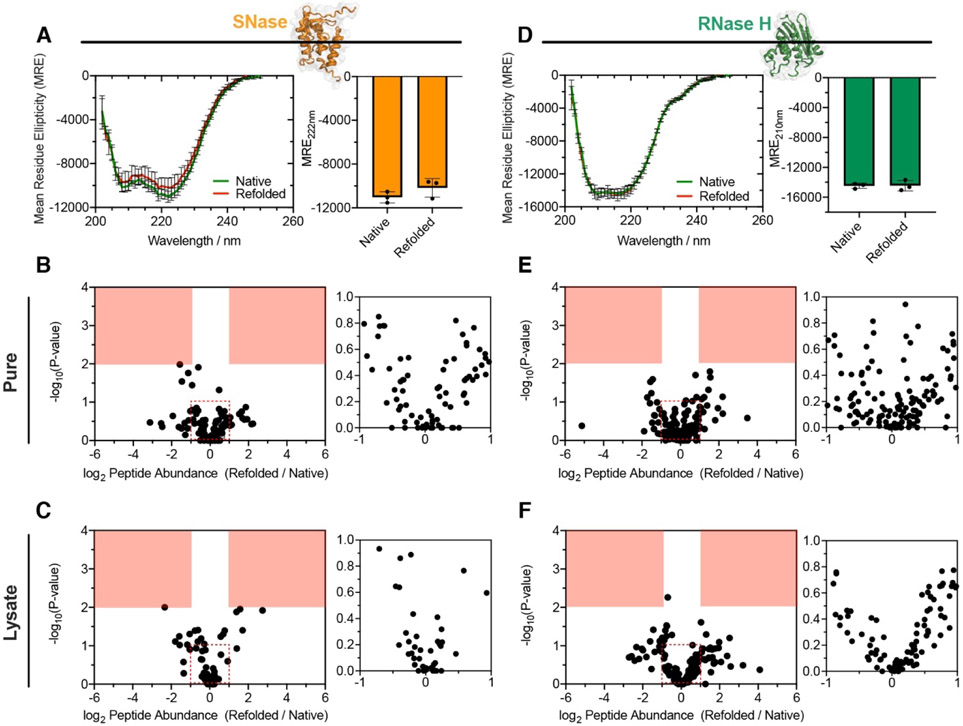
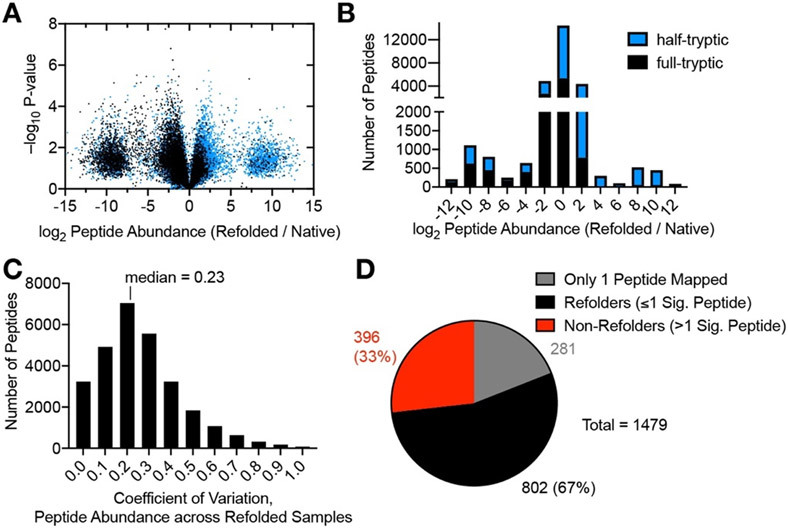
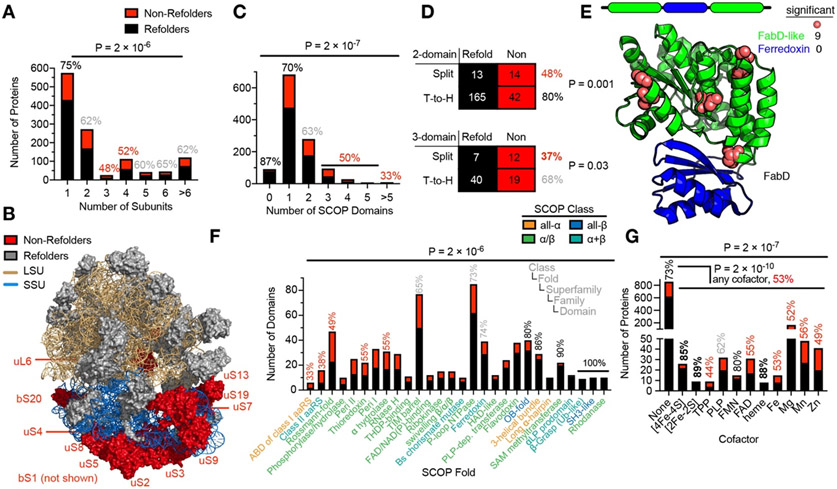
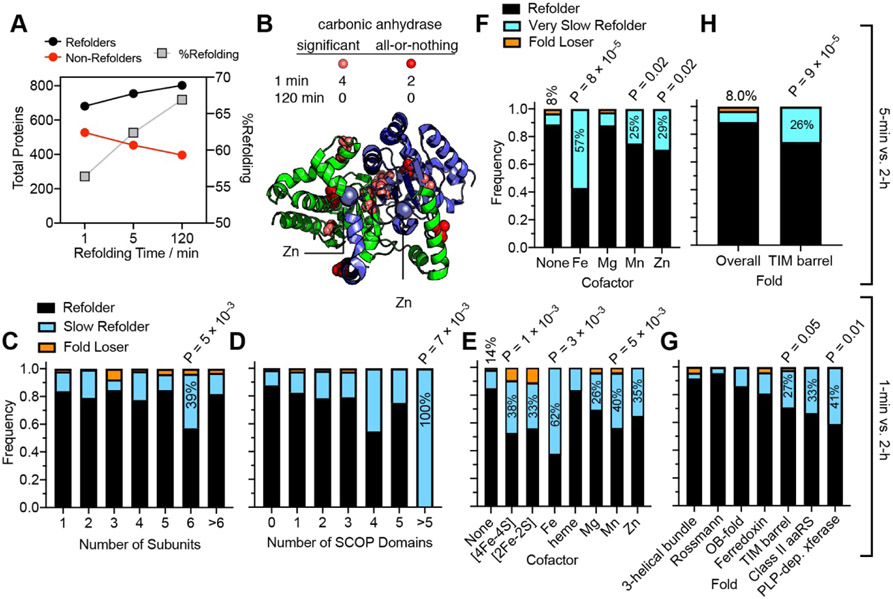
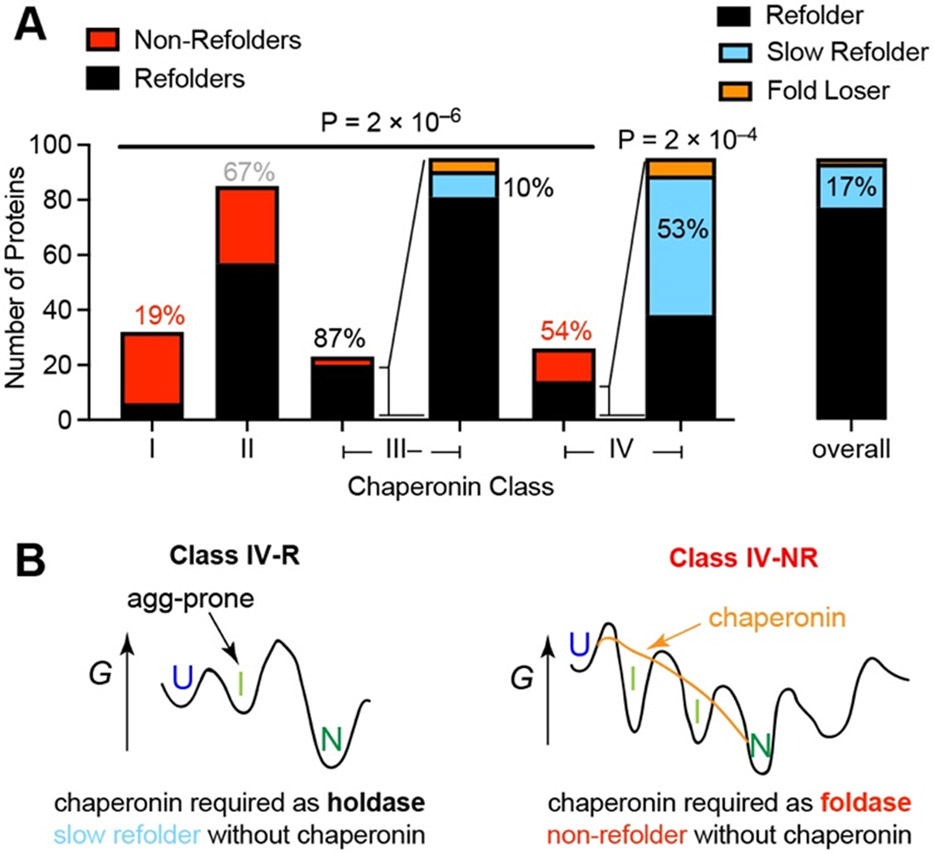
Decades of research on protein folding have primarily focused on a subset of small proteins that can reversibly refold from a denatured state. However, these studies have generally not been representative of the complexity of natural proteomes, which consist of many proteins with complex architectures and domain organizations. Here, we introduce an experimental approach to probe protein refolding kinetics for whole proteomes using mass spectrometry-based proteomics. Our study covers the majority of the soluble E. coli proteome expressed during log-phase growth, and among this group, we find that one-third of the E. coli proteome is not intrinsically refoldable on physiological time scales, a cohort that is enriched with certain fold-types, domain organizations, and other biophysical features. We also identify several properties and fold-types that are correlated with slow refolding on the minute time scale. Hence, these results illuminate when exogenous factors and processes, such as chaperones or cotranslational folding, might be required for efficient protein folding.
Figures
Similar articles
-
Folding the proteome.Trends Biochem Sci. 2013 Jul;38(7):337-44. doi: 10.1016/j.tibs.2013.05.001. Epub 2013 Jun 11. Trends Biochem Sci. 2013. PMID: 23764454 Free PMC article. Review.
-
The E. coli S30 lysate proteome: A prototype for cell-free protein production.N Biotechnol. 2018 Jan 25;40(Pt B):245-260. doi: 10.1016/j.nbt.2017.09.005. Epub 2017 Sep 21. N Biotechnol. 2018. PMID: 28943390
-
In vivo translation rates can substantially delay the cotranslational folding of the Escherichia coli cytosolic proteome.Proc Natl Acad Sci U S A. 2013 Jan 8;110(2):E132-40. doi: 10.1073/pnas.1213624110. Epub 2012 Dec 19. Proc Natl Acad Sci U S A. 2013. PMID: 23256155 Free PMC article.
-
Proteome folding kinetics is limited by protein halflife.PLoS One. 2014 Nov 13;9(11):e112701. doi: 10.1371/journal.pone.0112701. eCollection 2014. PLoS One. 2014. PMID: 25393560 Free PMC article.
-
What distinguishes GroEL substrates from other Escherichia coli proteins?FEBS J. 2012 Feb;279(4):543-50. doi: 10.1111/j.1742-4658.2011.08458.x. Epub 2012 Jan 4. FEBS J. 2012. PMID: 22177460 Review.
Cited by
-
FLiPPR: A Processor for Limited Proteolysis (LiP) Mass Spectrometry Datasets Built on FragPipe.bioRxiv [Preprint]. 2023 Dec 5:2023.12.04.569947. doi: 10.1101/2023.12.04.569947. bioRxiv. 2023. Update in: J Proteome Res. 2024 Jul 5;23(7):2332-2342. doi: 10.1021/acs.jproteome.3c00887. PMID: 38106106 Free PMC article. Updated. Preprint.
-
Protein surface chemistry encodes an adaptive tolerance to desiccation.bioRxiv [Preprint]. 2024 Oct 10:2024.07.28.604841. doi: 10.1101/2024.07.28.604841. bioRxiv. 2024. PMID: 39131385 Free PMC article. Preprint.
-
A proteome-wide map of chaperone-assisted protein refolding in a cytosol-like milieu.Proc Natl Acad Sci U S A. 2022 Nov 29;119(48):e2210536119. doi: 10.1073/pnas.2210536119. Epub 2022 Nov 23. Proc Natl Acad Sci U S A. 2022. PMID: 36417429 Free PMC article.
-
Peptides before and during the nucleotide world: an origins story emphasizing cooperation between proteins and nucleic acids.J R Soc Interface. 2022 Feb;19(187):20210641. doi: 10.1098/rsif.2021.0641. Epub 2022 Feb 9. J R Soc Interface. 2022. PMID: 35135297 Free PMC article. Review.
-
Non-Equilibrium Protein Folding and Activation by ATP-Driven Chaperones.Biomolecules. 2022 Jun 15;12(6):832. doi: 10.3390/biom12060832. Biomolecules. 2022. PMID: 35740957 Free PMC article.
References
-
- Dobson CM; Evans PA The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature 1992, 358, 302–307. - PubMed
-
- Hughson FM; Wright PE; Baldwin RL Structural Characterization of a Partly Folded Apomyoglobin Intermediate. Science 1990, 249, 1544–1548. - PubMed
-
- Lipman EA; Schuler B; Bakajin O; Eaton WA Single-Molecule Measurement of Protein Folding Kinetics. Science 2003, 301, 1233–1235. - PubMed
-
- Matouschek A; Kellis J; Serrano L; Fersht A Mapping the Transition State and Pathway of Protein Folding by Protein Engineering. Nature 1989, 340, 122–126. - PubMed
-
- Padmanabhan S; Marqusee S; Ridgeway T; Laue TM; Baldwin RL Relative Helix-Forming Tendencies of Nonpolar Amino Acids. Nature 1990, 344, 268–270. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
