

Stochastic pausing at latent HIV-1 promoters generates transcriptional bursting
- PMID: 34301927
- PMCID: PMC8302722
- DOI: 10.1038/s41467-021-24462-5
Stochastic pausing at latent HIV-1 promoters generates transcriptional bursting
Abstract
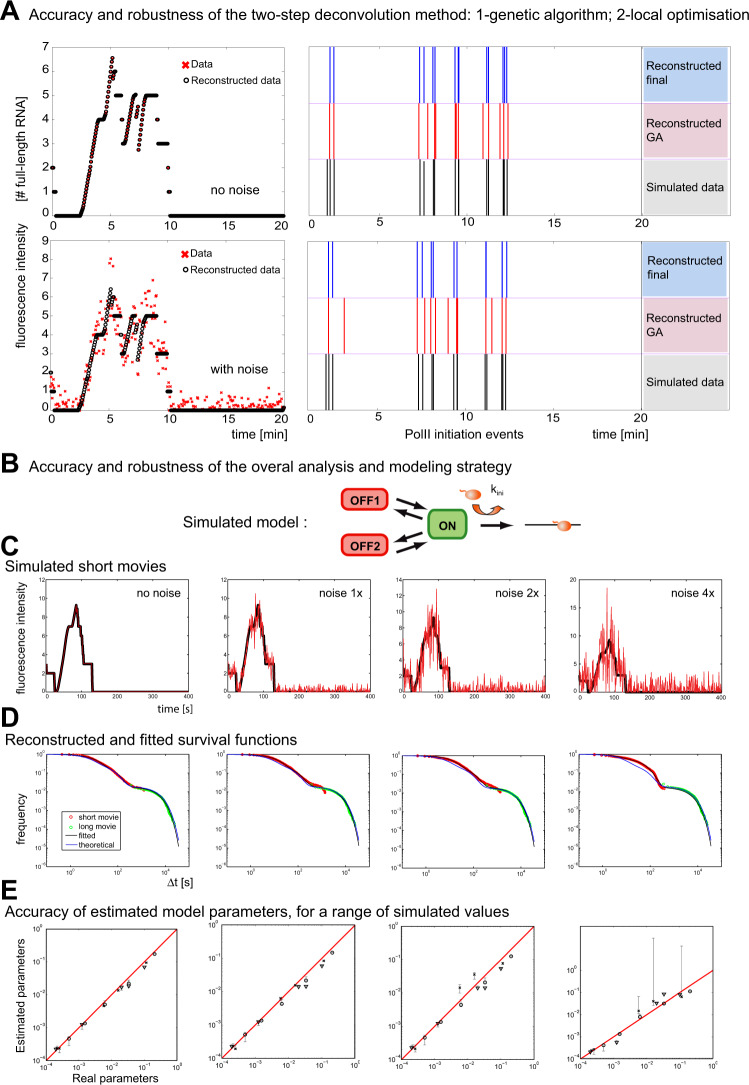
Promoter-proximal pausing of RNA polymerase II is a key process regulating gene expression. In latent HIV-1 cells, it prevents viral transcription and is essential for latency maintenance, while in acutely infected cells the viral factor Tat releases paused polymerase to induce viral expression. Pausing is fundamental for HIV-1, but how it contributes to bursting and stochastic viral reactivation is unclear. Here, we performed single molecule imaging of HIV-1 transcription. We developed a quantitative analysis method that manages multiple time scales from seconds to days and that rapidly fits many models of promoter dynamics. We found that RNA polymerases enter a long-lived pause at latent HIV-1 promoters (>20 minutes), thereby effectively limiting viral transcription. Surprisingly and in contrast to current models, pausing appears stochastic and not obligatory, with only a small fraction of the polymerases undergoing long-lived pausing in absence of Tat. One consequence of stochastic pausing is that HIV-1 transcription occurs in bursts in latent cells, thereby facilitating latency exit and providing a rationale for the stochasticity of viral rebounds.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







Similar articles
-
A Stronger Transcription Regulatory Circuit of HIV-1C Drives the Rapid Establishment of Latency with Implications for the Direct Involvement of Tat.J Virol. 2020 Sep 15;94(19):e00503-20. doi: 10.1128/JVI.00503-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32669338 Free PMC article.
-
Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat.Mol Cell Biol. 2014 Jun;34(11):1911-28. doi: 10.1128/MCB.01013-13. Epub 2014 Mar 17. Mol Cell Biol. 2014. PMID: 24636995 Free PMC article.
-
Posttranscriptional Regulation of HIV-1 Gene Expression during Replication and Reactivation from Latency by Nuclear Matrix Protein MATR3.mBio. 2018 Nov 13;9(6):e02158-18. doi: 10.1128/mBio.02158-18. mBio. 2018. PMID: 30425153 Free PMC article.
-
The Molecular Basis for Human Immunodeficiency Virus Latency.Annu Rev Virol. 2017 Sep 29;4(1):261-285. doi: 10.1146/annurev-virology-101416-041646. Epub 2017 Jul 17. Annu Rev Virol. 2017. PMID: 28715973 Review.
-
Selective recognition of viral promoters by host cell transcription complexes: challenges and opportunities to control latency.Curr Opin Virol. 2013 Aug;3(4):380-6. doi: 10.1016/j.coviro.2013.06.006. Epub 2013 Jul 1. Curr Opin Virol. 2013. PMID: 23827503 Review.
Cited by
-
Viro-fluidics: Real-time analysis of virus production kinetics at the single-cell level.Biophys Rep (N Y). 2022 Aug 11;2(3):100068. doi: 10.1016/j.bpr.2022.100068. eCollection 2022 Sep 14. Biophys Rep (N Y). 2022. PMID: 36425325 Free PMC article.
-
BurstDECONV: a signal deconvolution method to uncover mechanisms of transcriptional bursting in live cells.Nucleic Acids Res. 2023 Sep 8;51(16):e88. doi: 10.1093/nar/gkad629. Nucleic Acids Res. 2023. PMID: 37522372 Free PMC article.
-
A Two-Color Haploid Genetic Screen Identifies Novel Host Factors Involved in HIV-1 Latency.mBio. 2021 Dec 21;12(6):e0298021. doi: 10.1128/mBio.02980-21. Epub 2021 Dec 7. mBio. 2021. PMID: 34872356 Free PMC article.
-
Genome-wide inference reveals that feedback regulations constrain promoter-dependent transcriptional burst kinetics.Nucleic Acids Res. 2023 Jan 11;51(1):68-83. doi: 10.1093/nar/gkac1204. Nucleic Acids Res. 2023. PMID: 36583343 Free PMC article.
-
Release of P-TEFb from the Super Elongation Complex promotes HIV-1 latency reversal.PLoS Pathog. 2024 Sep 11;20(9):e1012083. doi: 10.1371/journal.ppat.1012083. eCollection 2024 Sep. PLoS Pathog. 2024. PMID: 39259751 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
