

Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16
- PMID: 34070858
- PMCID: PMC8199271
- DOI: 10.3390/ijms22115870
Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16
Abstract
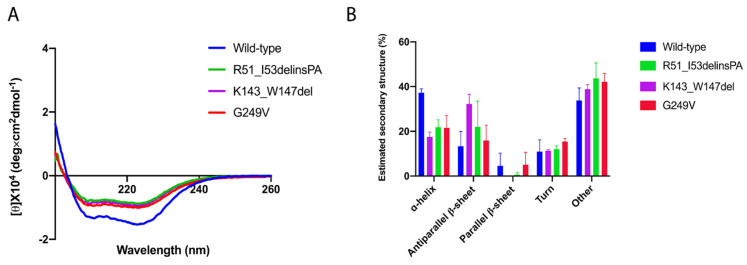
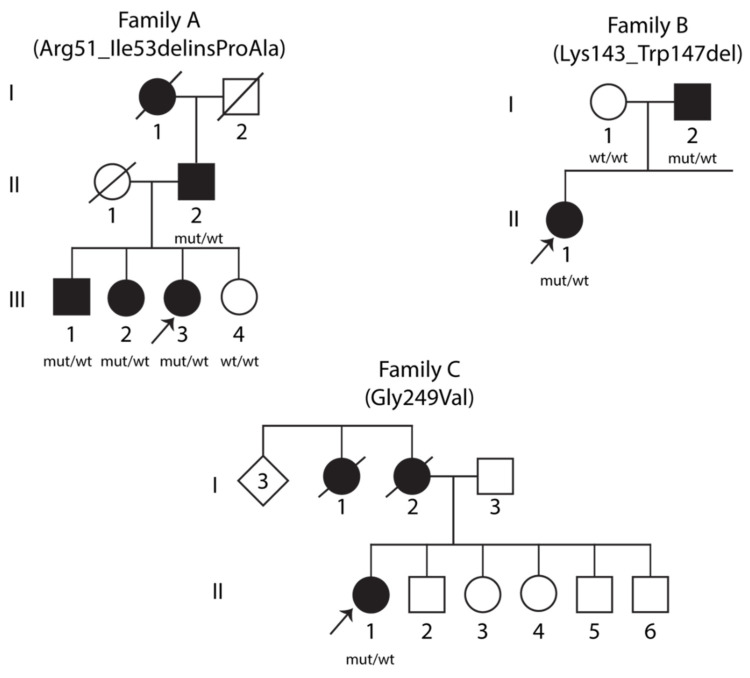
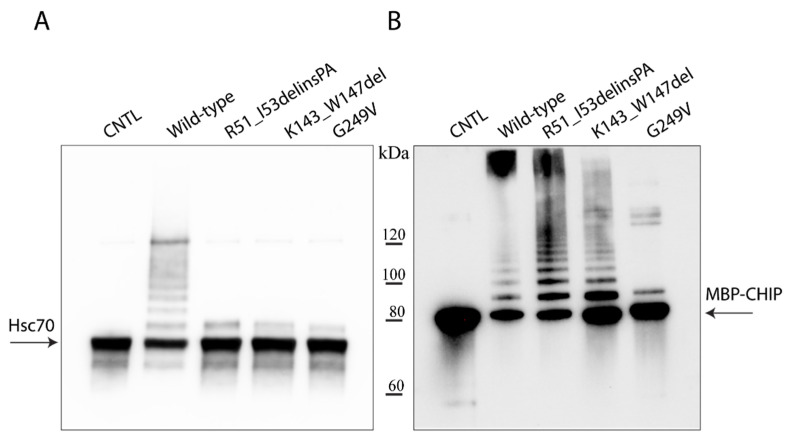
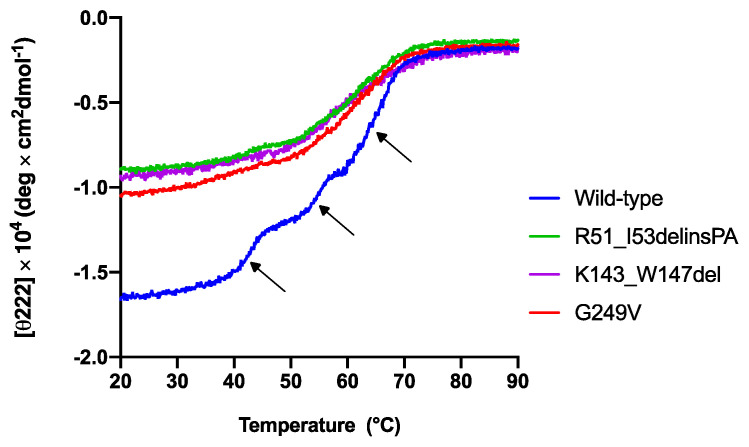
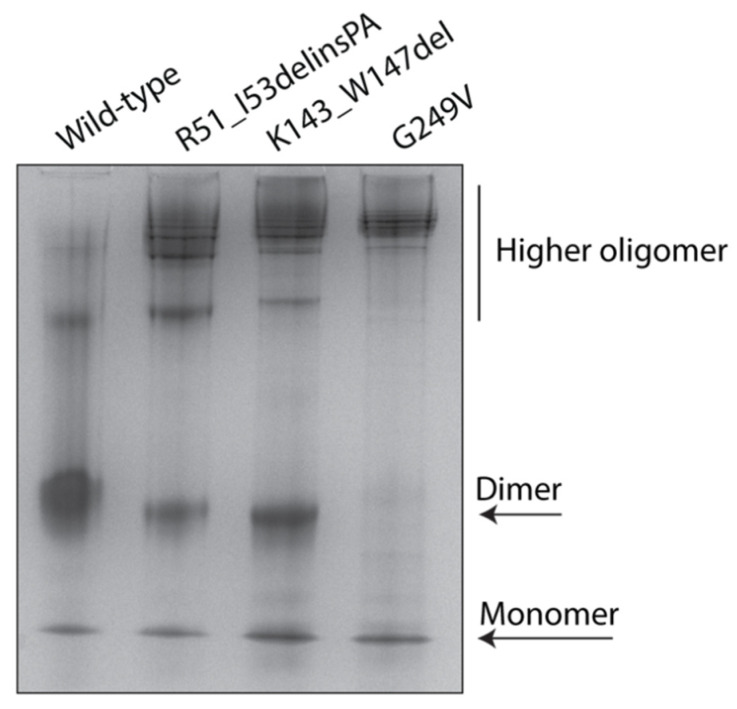
Variants in STUB1 cause both autosomal recessive (SCAR16) and dominant (SCA48) spinocerebellar ataxia. Reports from 18 STUB1 variants causing SCA48 show that the clinical picture includes later-onset ataxia with a cerebellar cognitive affective syndrome and varying clinical overlap with SCAR16. However, little is known about the molecular properties of dominant STUB1 variants. Here, we describe three SCA48 families with novel, dominantly inherited STUB1 variants (p.Arg51_Ile53delinsProAla, p.Lys143_Trp147del, and p.Gly249Val). All the patients developed symptoms from 30 years of age or later, all had cerebellar atrophy, and 4 had cognitive/psychiatric phenotypes. Investigation of the structural and functional consequences of the recombinant C-terminus of HSC70-interacting protein (CHIP) variants was performed in vitro using ubiquitin ligase activity assay, circular dichroism assay and native polyacrylamide gel electrophoresis. These studies revealed that dominantly and recessively inherited STUB1 variants showed similar biochemical defects, including impaired ubiquitin ligase activity and altered oligomerization properties of the CHIP. Our findings expand the molecular understanding of SCA48 but also mean that assumptions concerning unaffected carriers of recessive STUB1 variants in SCAR16 families must be re-evaluated. More investigations are needed to verify the disease status of SCAR16 heterozygotes and elucidate the molecular relationship between SCA48 and SCAR16 diseases.
Keywords: CHIP; E3 ubiquitin ligase; SCA48; SCAR16; STUB1; spinocerebellar ataxia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Spinocerebellar ataxia type 48: last but not least.Neurol Sci. 2020 Sep;41(9):2423-2432. doi: 10.1007/s10072-020-04408-3. Epub 2020 Apr 27. Neurol Sci. 2020. PMID: 32342324 Review.
-
Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48).Neurology. 2018 Nov 20;91(21):e1988-e1998. doi: 10.1212/WNL.0000000000006550. Epub 2018 Oct 31. Neurology. 2018. PMID: 30381368
-
The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families.Eur J Neurol. 2020 Mar;27(3):498-505. doi: 10.1111/ene.14094. Epub 2019 Nov 1. Eur J Neurol. 2020. PMID: 31571321
-
Clinical and functional characterization of a novel STUB1 mutation in a Chinese spinocerebellar ataxia 48 pedigree.Orphanet J Rare Dis. 2024 Dec 20;19(1):471. doi: 10.1186/s13023-024-03456-8. Orphanet J Rare Dis. 2024. PMID: 39707479 Free PMC article.
-
Autosomal dominant cerebellar ataxias: new genes and progress towards treatments.Lancet Neurol. 2023 Aug;22(8):735-749. doi: 10.1016/S1474-4422(23)00068-6. Lancet Neurol. 2023. PMID: 37479376 Review.
Cited by
-
Late-onset sensory-motor axonal neuropathy, a novel SLC12A6-related phenotype.Brain. 2023 Mar 1;146(3):912-922. doi: 10.1093/brain/awac488. Brain. 2023. PMID: 36542484 Free PMC article.
-
Mutations in Hsp90 Cochaperones Result in a Wide Variety of Human Disorders.Front Mol Biosci. 2021 Dec 8;8:787260. doi: 10.3389/fmolb.2021.787260. eCollection 2021. Front Mol Biosci. 2021. PMID: 34957217 Free PMC article. Review.
-
Chip Protein U-Box Domain Truncation Affects Purkinje Neuron Morphology and Leads to Behavioral Changes in Zebrafish.Front Mol Neurosci. 2021 Sep 24;14:723912. doi: 10.3389/fnmol.2021.723912. eCollection 2021. Front Mol Neurosci. 2021. PMID: 34630034 Free PMC article.
-
The molecular basis of spinocerebellar ataxia type 48 caused by a de novo mutation in the ubiquitin ligase CHIP.J Biol Chem. 2022 May;298(5):101899. doi: 10.1016/j.jbc.2022.101899. Epub 2022 Apr 7. J Biol Chem. 2022. PMID: 35398354 Free PMC article.
-
MRI Findings in a Patient with Known SCAR-16 Type STUB1 Associated Cerebellar Ataxia.J Belg Soc Radiol. 2022 Dec 14;106(1):131. doi: 10.5334/jbsr.2902. eCollection 2022. J Belg Soc Radiol. 2022. PMID: 36569391 Free PMC article.
References
-
- Bird T.D. Hereditary Ataxia Overview. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. GeneReviews(®) University of Washington; Washington, WA, USA: 1993. - PubMed
-
- Genis D., Ortega-Cubero S., San Nicolás H., Corral J., Gardenyes J., de Jorge L., López E., Campos B., Lorenzo E., Tonda R., et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48) Neurology. 2018;91:e1988–e1998. doi: 10.1212/WNL.0000000000006550. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
