

A BMPR2/YY1 Signaling Axis Is Required for Human Cytomegalovirus Latency in Undifferentiated Myeloid Cells
- PMID: 34061599
- PMCID: PMC8262994
- DOI: 10.1128/mBio.00227-21
A BMPR2/YY1 Signaling Axis Is Required for Human Cytomegalovirus Latency in Undifferentiated Myeloid Cells
Abstract
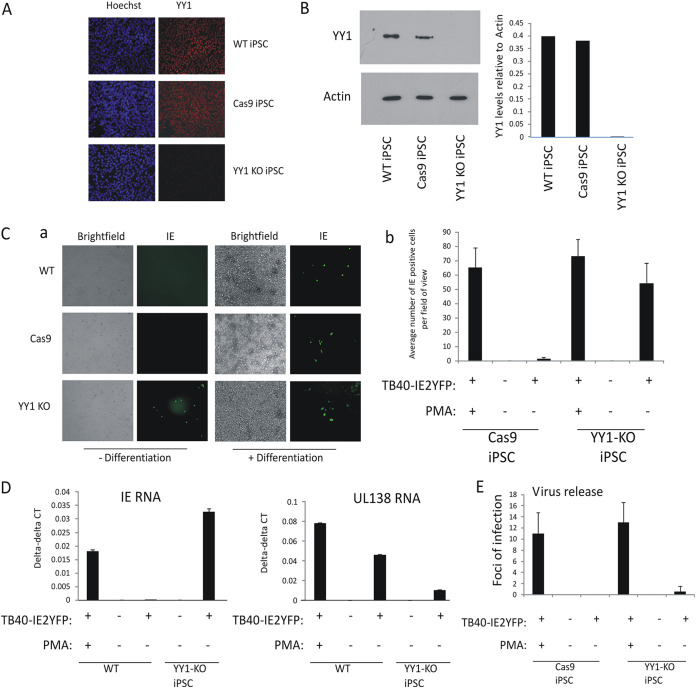
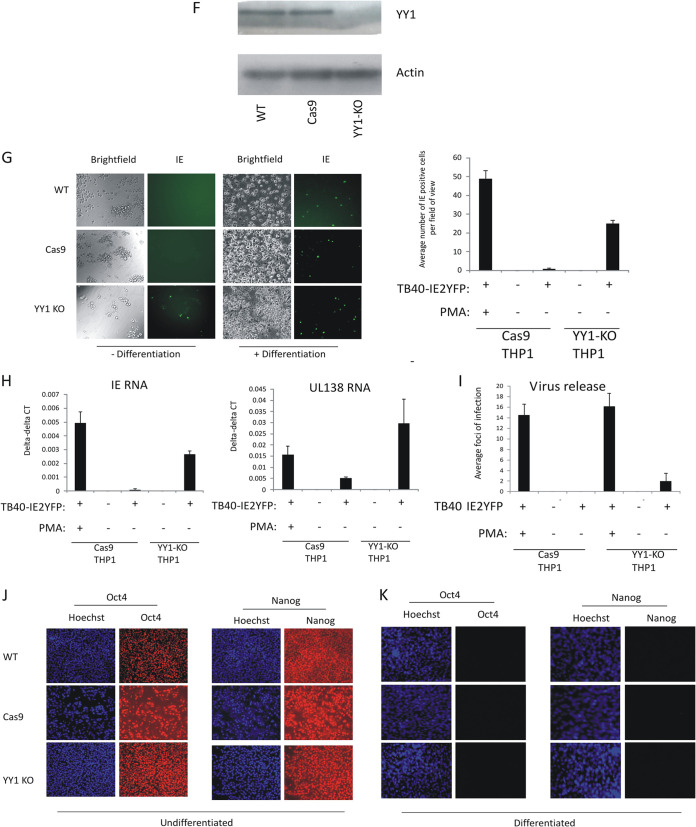
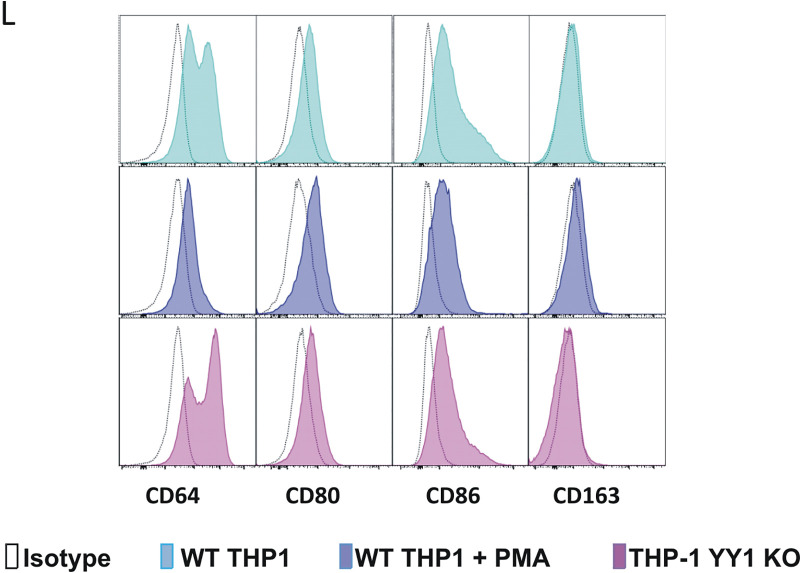
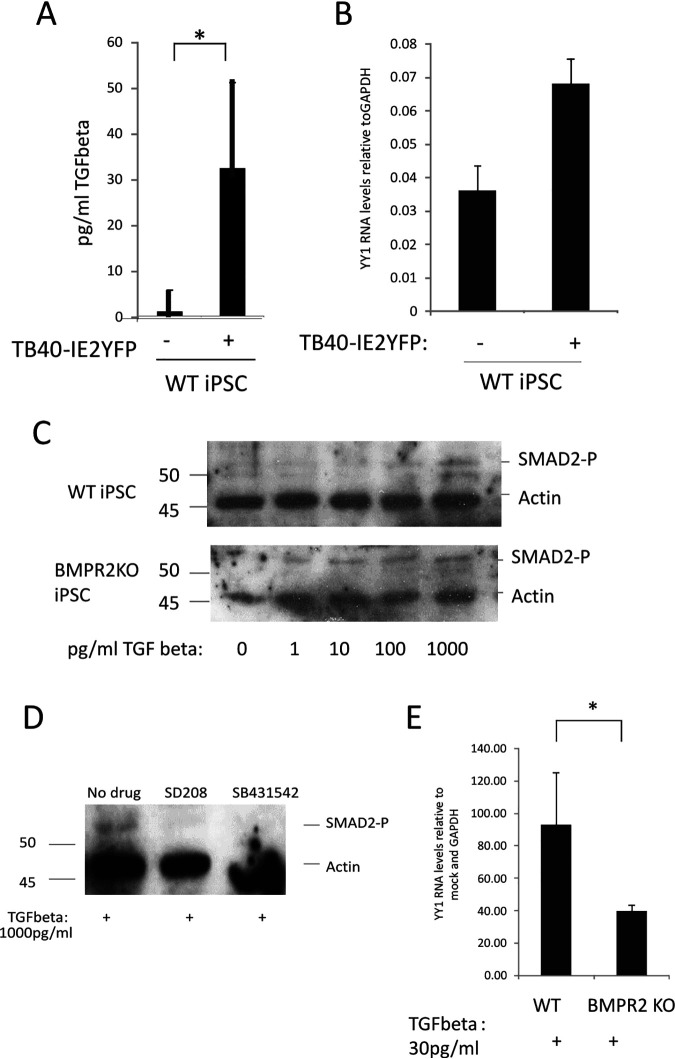
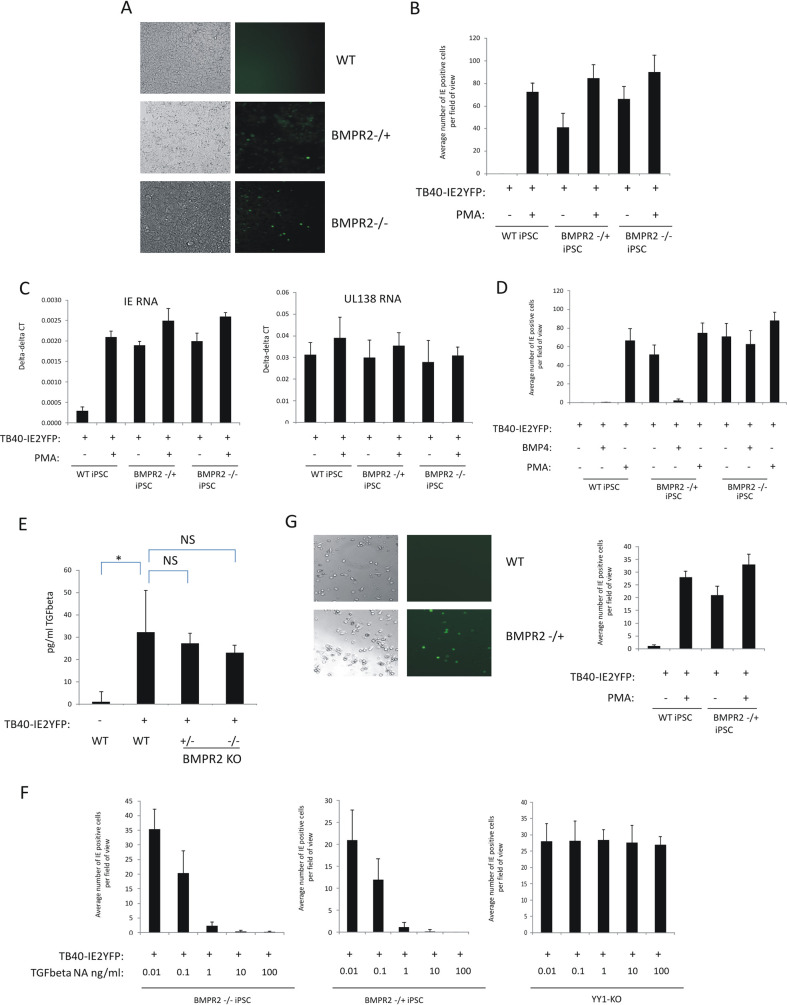
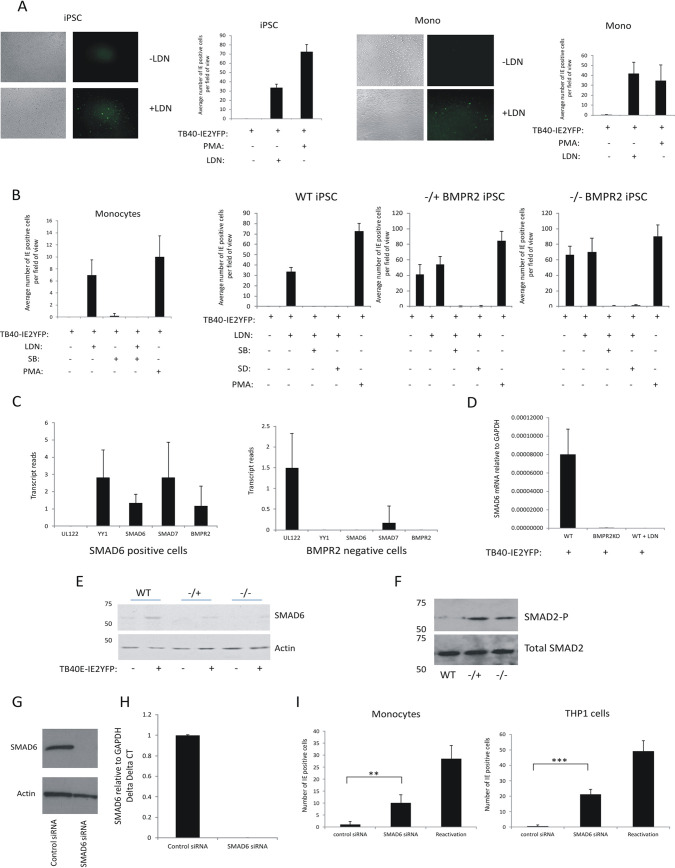
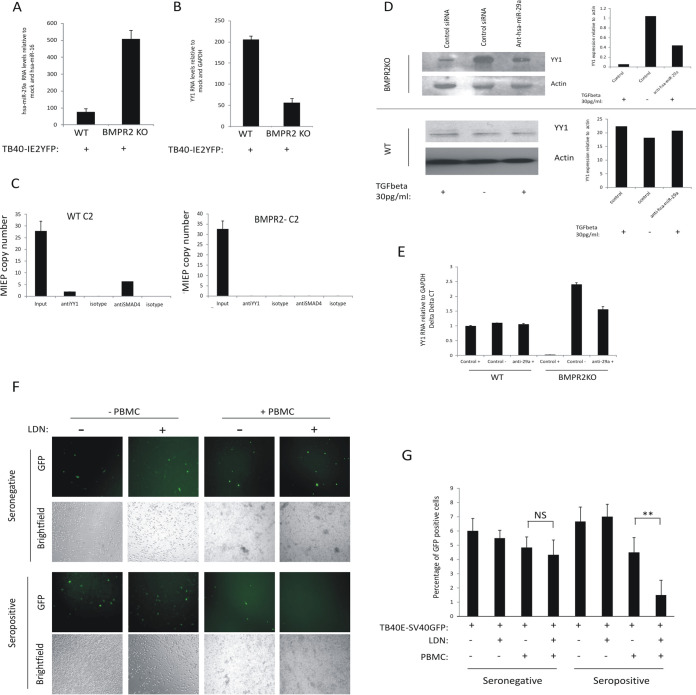
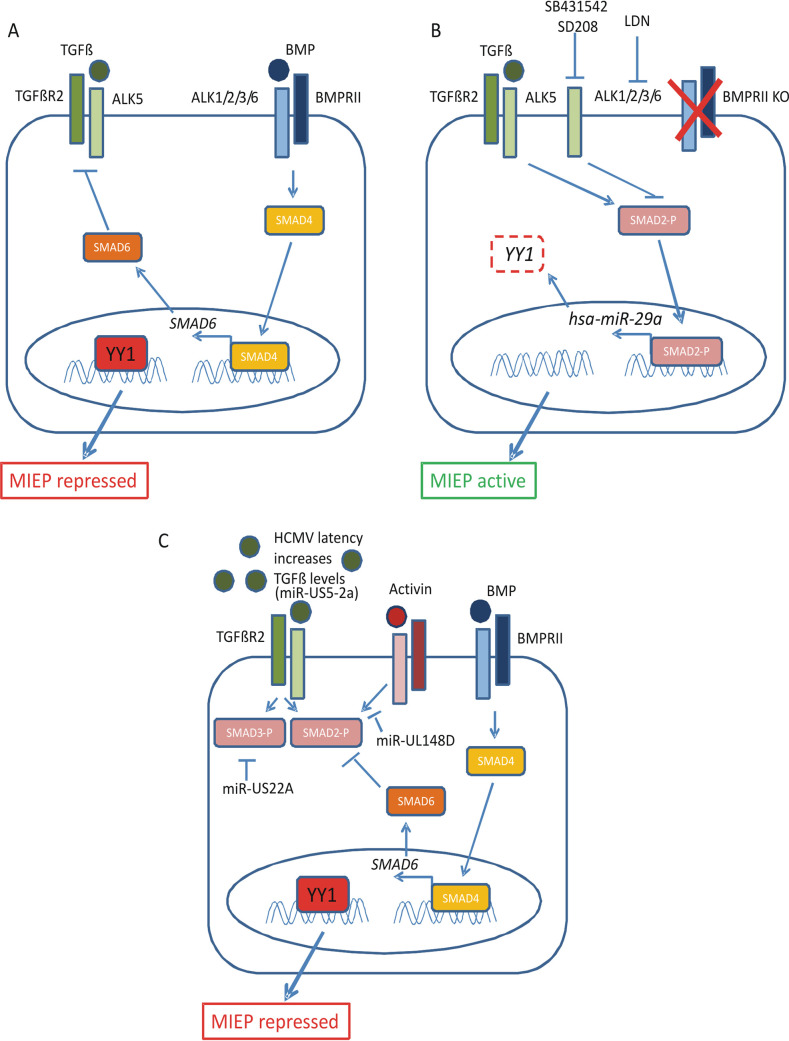
Human cytomegalovirus (HCMV) presents a major health burden in the immunocompromised and in stem cell transplant medicine. A lack of understanding about the mechanisms of HCMV latency in undifferentiated CD34+ stem cells, and how latency is broken for the virus to enter the lytic phase of its infective cycle, has hampered the development of essential therapeutics. Using a human induced pluripotent stem cell (iPSC) model of HCMV latency and patient-derived myeloid cell progenitors, we demonstrate that bone morphogenetic protein receptor type 2 (BMPR2) is necessary for HCMV latency. In addition, we define a crucial role for the transcription factor Yin Yang 1 (YY1) in HCMV latency; high levels of YY1 are maintained in latently infected cells as a result of BMPR2 signaling through the SMAD4/SMAD6 axis. Activation of SMAD4/6, through BMPR2, inhibits TGFbeta receptor signaling, which leads to the degradation of YY1 via induction of a cellular microRNA (miRNA), hsa-miR-29a. Pharmacological targeting of BMPR2 in progenitor cells results in the degradation of YY1 and an inability to maintain latency and renders cells susceptible to T cell killing. These data argue that BMPR2 plays a role in HCMV latency and is a new potential therapeutic target for maintaining or disrupting HCMV latency in myeloid progenitors. IMPORTANCE Understanding the mechanisms which regulate HCMV latency could allow therapeutic targeting of the latent virus reservoir from where virus reactivation can cause severe disease. We show that the BMPR2/TGFbeta receptor/YY1 signaling axis is crucial to maintain HCMV latency in undifferentiated cells and that pharmacological reduction of BMPR2 in latently infected cells leads to reactivation of the viral lytic transcription program, which renders the infected cell open to immune detection and clearance in infected individuals. Therefore, this work identifies key host-virus interactions which regulate HCMV latent infection. It also demonstrates a potential new therapeutic approach to reduce HCMV reactivation-mediated disease by the treatment of donor stem cells/organs prior to transplantation, which could have a major impact in the transplant disease setting.
Keywords: BMPR2; YY1; human cytomegalovirus; latency; stem cells.
Figures
Similar articles
-
Human Cytomegalovirus US28 Ligand Binding Activity Is Required for Latency in CD34+ Hematopoietic Progenitor Cells and Humanized NSG Mice.mBio. 2019 Aug 20;10(4):e01889-19. doi: 10.1128/mBio.01889-19. mBio. 2019. PMID: 31431555 Free PMC article.
-
Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways To Maintain Latent Infection.mBio. 2017 Dec 5;8(6):e01754-17. doi: 10.1128/mBio.01754-17. mBio. 2017. PMID: 29208743 Free PMC article.
-
Human Cytomegalovirus Requires Epidermal Growth Factor Receptor Signaling To Enter and Initiate the Early Steps in the Establishment of Latency in CD34+ Human Progenitor Cells.J Virol. 2017 Feb 14;91(5):e01206-16. doi: 10.1128/JVI.01206-16. Print 2017 Mar 1. J Virol. 2017. PMID: 27974567 Free PMC article.
-
Aspects of human cytomegalovirus latency and reactivation.Curr Top Microbiol Immunol. 2008;325:297-313. doi: 10.1007/978-3-540-77349-8_17. Curr Top Microbiol Immunol. 2008. PMID: 18637513 Review.
-
Human cytomegalovirus: Latency and reactivation in the myeloid lineage.J Clin Virol. 2008 Mar;41(3):180-5. doi: 10.1016/j.jcv.2007.11.014. J Clin Virol. 2008. PMID: 18164651 Review.
Cited by
-
Advances in Model Systems for Human Cytomegalovirus Latency and Reactivation.mBio. 2022 Feb 22;13(1):e0172421. doi: 10.1128/mbio.01724-21. Epub 2022 Jan 11. mBio. 2022. PMID: 35012351 Free PMC article. Review.
-
Epstein-Barr virus protein EBNA-LP engages YY1 through leucine-rich motifs to promote naïve B cell transformation.PLoS Pathog. 2024 Jul 31;20(7):e1011950. doi: 10.1371/journal.ppat.1011950. eCollection 2024 Jul. PLoS Pathog. 2024. PMID: 39083560 Free PMC article.
-
Latency-associated upregulation of SERBP1 is important for the recruitment of transcriptional repressors to the viral major immediate early promoter of human cytomegalovirus during latent carriage.Front Microbiol. 2022 Nov 24;13:999290. doi: 10.3389/fmicb.2022.999290. eCollection 2022. Front Microbiol. 2022. PMID: 36504797 Free PMC article.
-
The Human Cytomegalovirus Latency-Associated Gene Product Latency Unique Natural Antigen Regulates Latent Gene Expression.Viruses. 2023 Sep 4;15(9):1875. doi: 10.3390/v15091875. Viruses. 2023. PMID: 37766281 Free PMC article.
-
Modulation of host cell signaling during cytomegalovirus latency and reactivation.Virol J. 2021 Oct 18;18(1):207. doi: 10.1186/s12985-021-01674-1. Virol J. 2021. PMID: 34663377 Free PMC article. Review.
References
-
- Autmizguine J, Theoret Y, Launay E, Duval M, Rousseau C, Tapiero B, Boivin G, Ovetchkine P. 2011. Low systemic ganciclovir exposure and preemptive treatment failure of cytomegalovirus reactivation in a transplanted child. J Popul Ther Clin Pharmacol 18:e257–e260. - PubMed
-
- Sinclair J, Poole E. 2014. Human cytomegalovirus latency and reactivation in and beyond the myeloid lineage. Future Virol 9:557–563. doi:10.2217/fvl.14.34. - DOI
-
- Poole E, Wills M, Sinclair J. 2014. Human cytomegalovirus latency: targeting differences in the latently infected cell with a view to clearing latent infection. New J Sci 2014:1–10. doi:10.1155/2014/313761. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
