

ALK inhibition activates LC3B-independent, protective autophagy in EML4-ALK positive lung cancer cells
- PMID: 33907223
- PMCID: PMC8079437
- DOI: 10.1038/s41598-021-87966-6
ALK inhibition activates LC3B-independent, protective autophagy in EML4-ALK positive lung cancer cells
Abstract
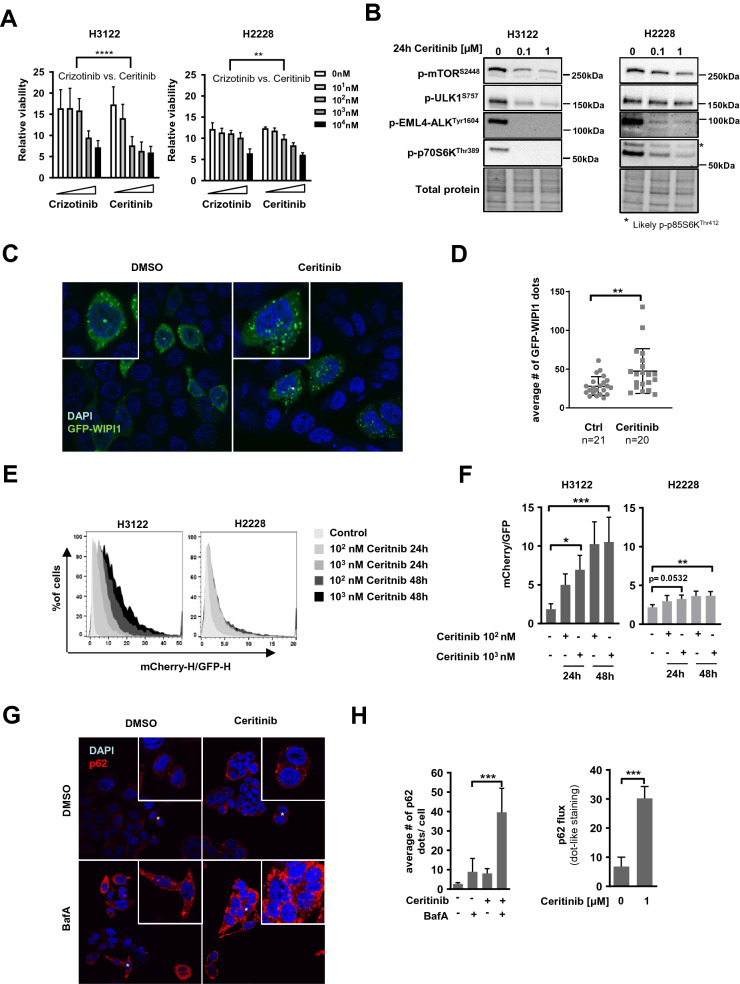
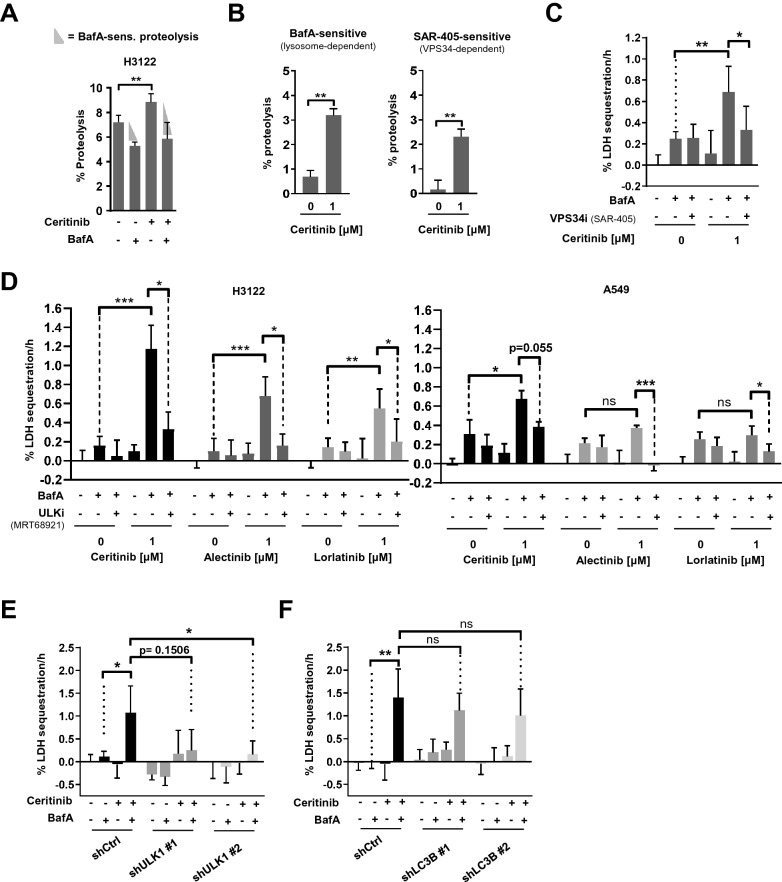
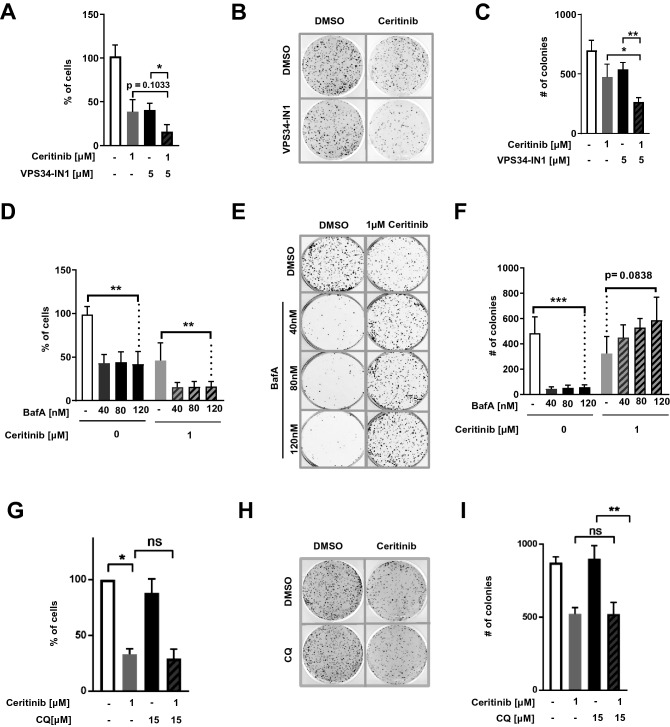
ALK inhibitors effectively target EML4-ALK positive non-small cell lung cancer, but their effects are hampered by treatment resistance. In the present study, we asked whether ALK inhibition affects autophagy, and whether this may influence treatment response. Whereas the impact of targeted therapies on autophagic activity previously have been assessed by surrogate marker proteins such as LC3B, we here thoroughly examined effects on functional autophagic activity, i.e. on the sequestration and degradation of autophagic cargo, in addition to autophagic markers. Interestingly, the ALK inhibitor Ceritinib decreased mTOR activity and increased GFP-WIPI1 dot formation in H3122 and H2228 EML4-ALK+ lung cancer cells, suggesting autophagy activation. Moreover, an mCherry-EGFP-LC3B based assay indicated elevated LC3B carrier flux upon ALK inhibition. In accordance, autophagic cargo sequestration and long-lived protein degradation significantly increased upon ALK inhibition. Intriguingly, autophagic cargo flux was dependent on VPS34 and ULK1, but not LC3B. Co-treating H3122 cells with Ceritinib and a VPS34 inhibitor or Bafilomycin A1 resulted in reduced cell numbers. Moreover, VPS34 inhibition reduced clonogenic recovery of Ceritinib-treated cells. In summary, our results indicate that ALK inhibition triggers LC3B-independent macroautophagic flux in EML4-ALK+ cells to support cancer cell survival and clonogenic growth.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer.Clin Cancer Res. 2012 Nov 15;18(22):6219-26. doi: 10.1158/1078-0432.CCR-12-0392. Epub 2012 Jul 27. Clin Cancer Res. 2012. PMID: 22843788
-
Oncogene addiction and radiation oncology: effect of radiotherapy with photons and carbon ions in ALK-EML4 translocated NSCLC.Radiat Oncol. 2018 Jan 5;13(1):1. doi: 10.1186/s13014-017-0947-0. Radiat Oncol. 2018. PMID: 29304828 Free PMC article.
-
A new human lung adenocarcinoma cell line harboring the EML4-ALK fusion gene.Jpn J Clin Oncol. 2014 Oct;44(10):963-8. doi: 10.1093/jjco/hyu110. Epub 2014 Aug 28. Jpn J Clin Oncol. 2014. PMID: 25170107
-
Role of anaplastic lymphoma kinase inhibition in the treatment of non-small-cell lung cancer.Am J Health Syst Pharm. 2015 Sep 1;72(17):1456-62. doi: 10.2146/ajhp140836. Am J Health Syst Pharm. 2015. PMID: 26294238 Review.
-
Ceritinib for Untreated Anaplastic Lymphoma Kinase-Positive Advanced Non-Small-Cell Lung Cancer: An Evidence Review Group Evaluation of a NICE Single Technology Appraisal.Pharmacoeconomics. 2019 May;37(5):645-654. doi: 10.1007/s40273-018-0720-8. Pharmacoeconomics. 2019. PMID: 30298279 Review.
Cited by
-
DSTYK inhibition increases the sensitivity of lung cancer cells to T cell-mediated cytotoxicity.J Exp Med. 2022 Dec 5;219(12):e20220726. doi: 10.1084/jem.20220726. Epub 2022 Sep 28. J Exp Med. 2022. PMID: 36169652 Free PMC article.
-
Novel Approach to Skin Anti-Aging: Boosting Pharmacological Effects of Exogenous Nicotinamide Adenine Dinucleotide (NAD+) by Synergistic Inhibition of CD38 Expression.Cells. 2024 Oct 30;13(21):1799. doi: 10.3390/cells13211799. Cells. 2024. PMID: 39513906 Free PMC article.
-
Perturbation of Cellular Redox Homeostasis Dictates Divergent Effects of Polybutyl Cyanoacrylate (PBCA) Nanoparticles on Autophagy.Cells. 2021 Dec 6;10(12):3432. doi: 10.3390/cells10123432. Cells. 2021. PMID: 34943939 Free PMC article.
-
The Role of Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3 in the Pathogenesis of Human Cancer.Int J Mol Sci. 2021 Oct 11;22(20):10964. doi: 10.3390/ijms222010964. Int J Mol Sci. 2021. PMID: 34681622 Free PMC article. Review.
-
Inhibition of autophagy; an opportunity for the treatment of cancer resistance.Front Cell Dev Biol. 2023 Jun 9;11:1177440. doi: 10.3389/fcell.2023.1177440. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37363731 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
