

Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19
- PMID: 33713619
- PMCID: PMC7874909
- DOI: 10.1016/j.cell.2021.02.018
Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19
Abstract
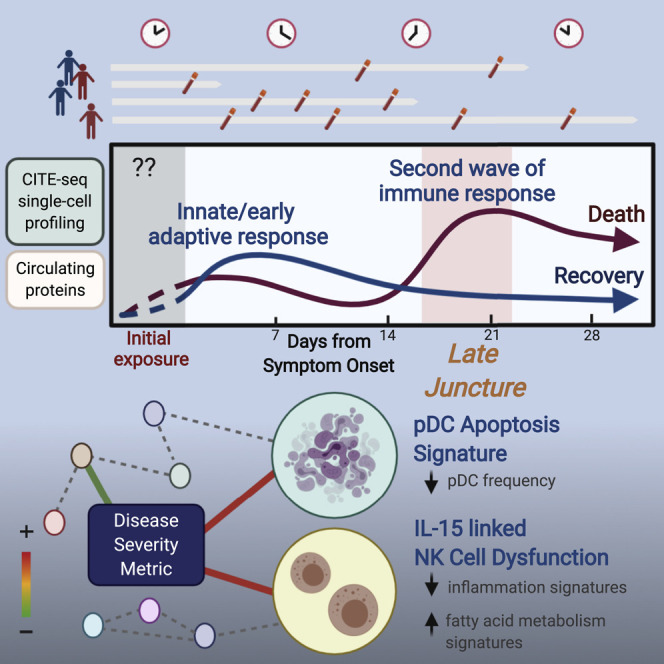
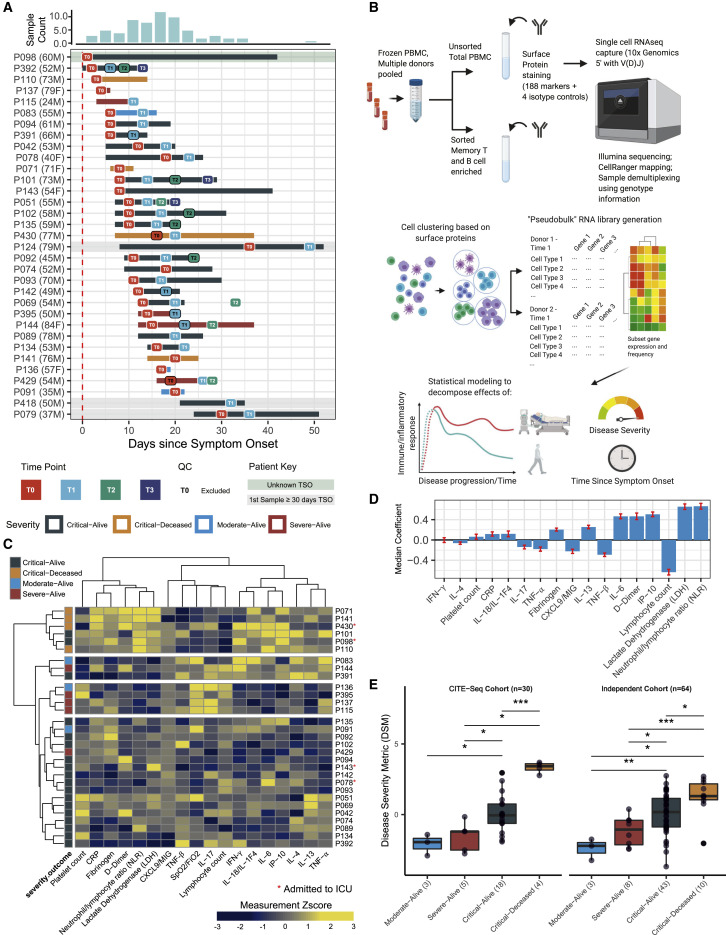
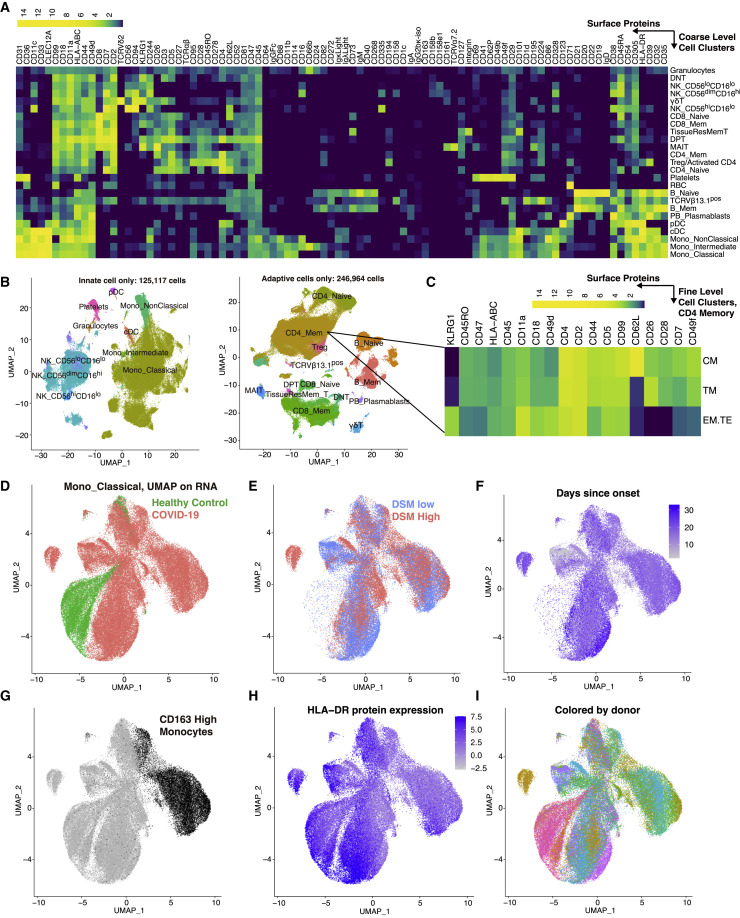
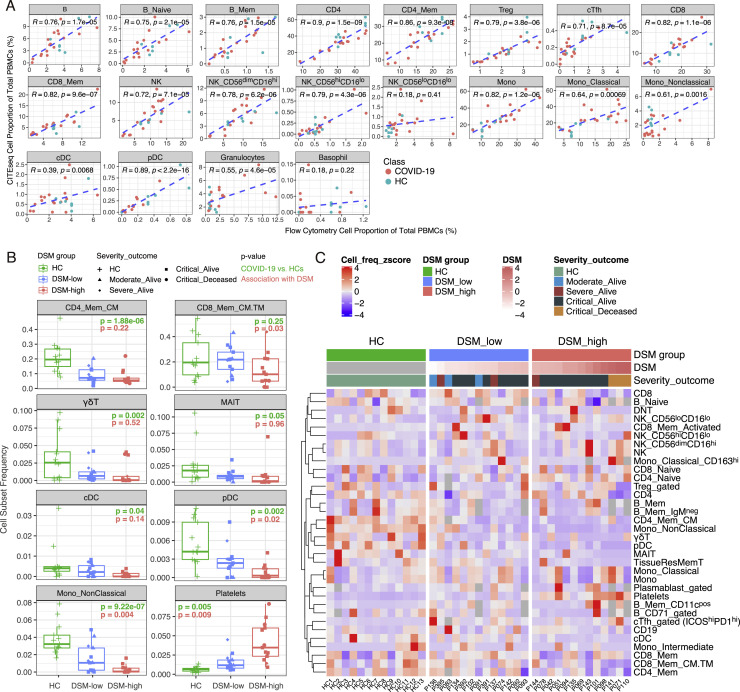
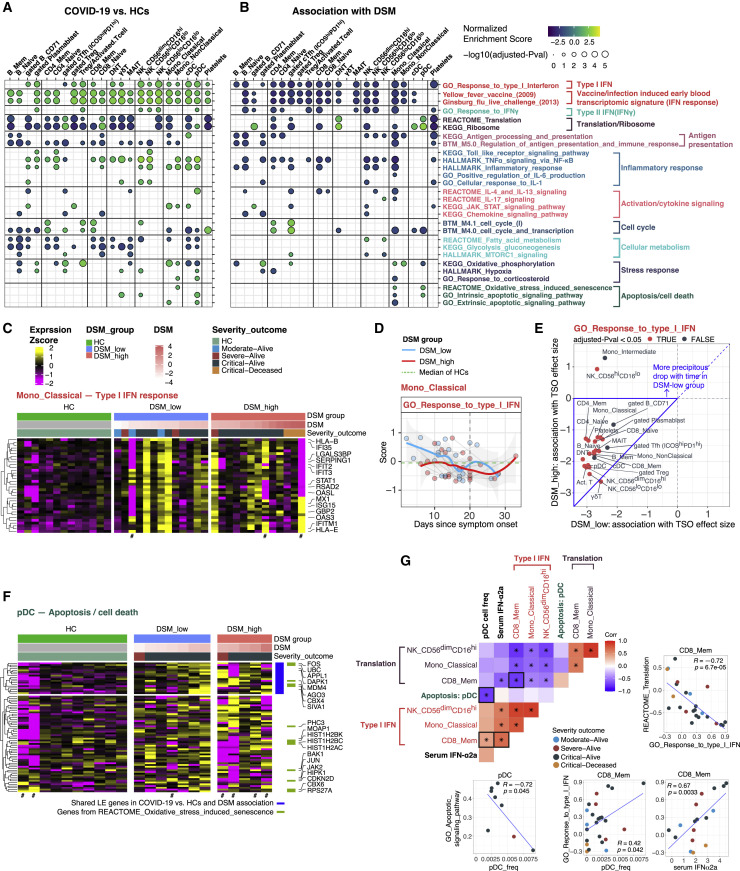
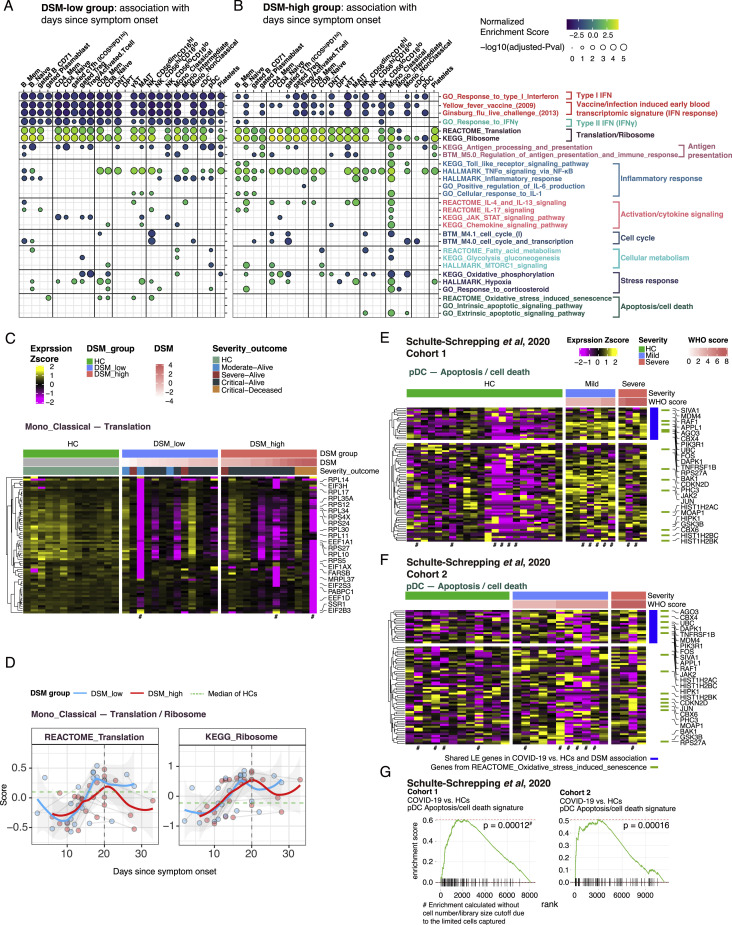
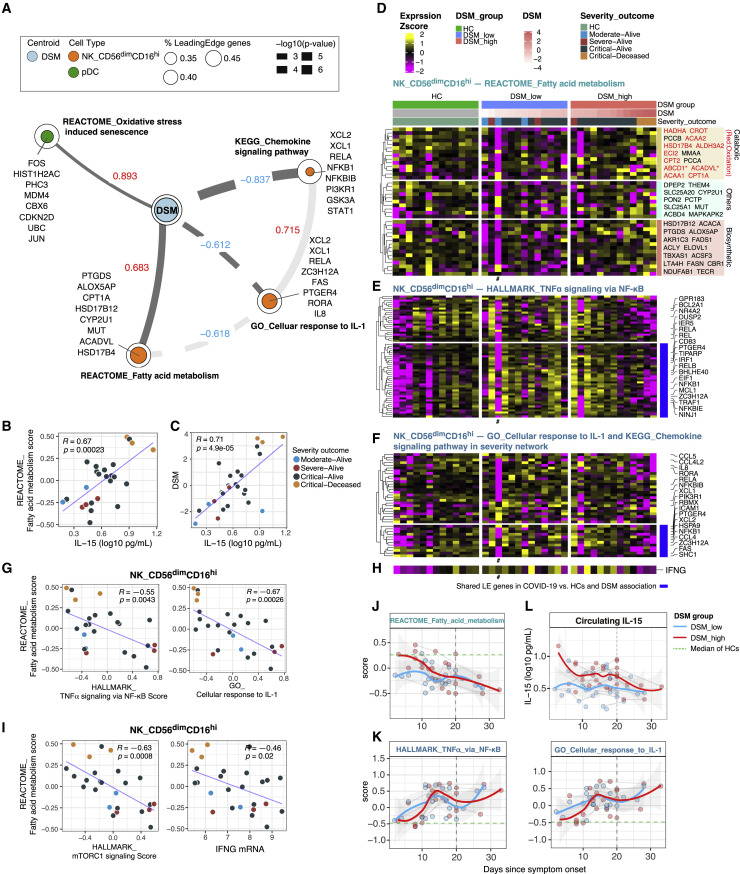
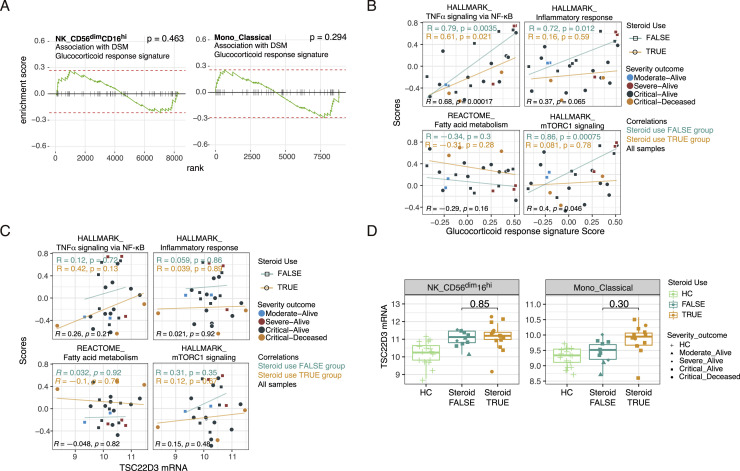
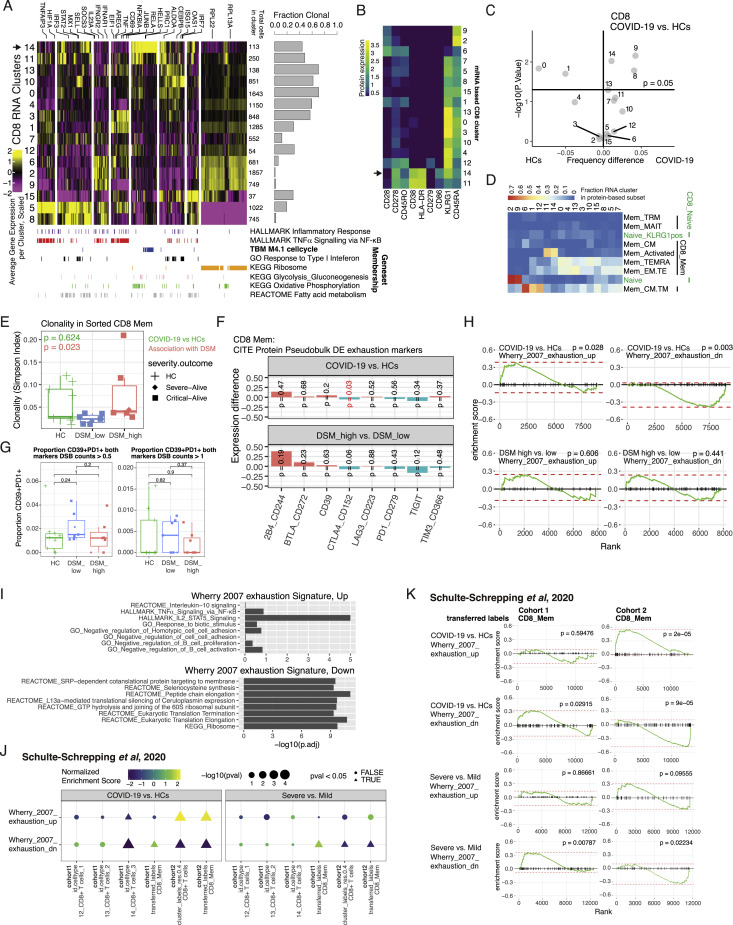
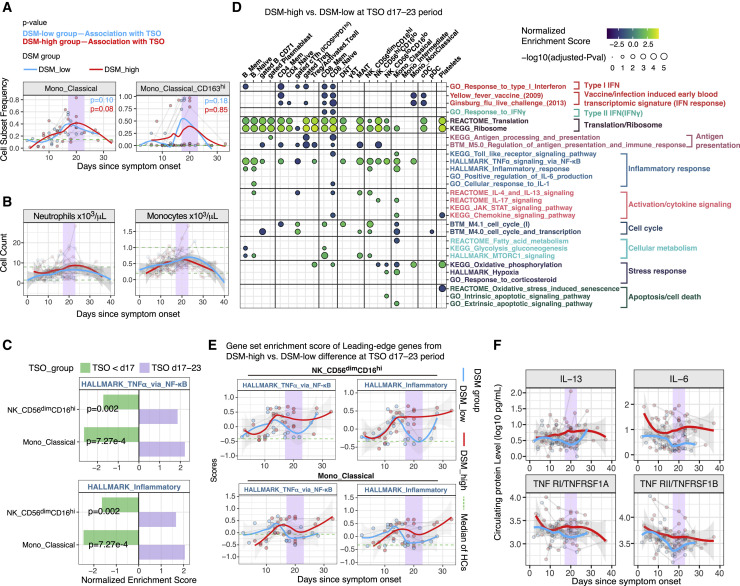
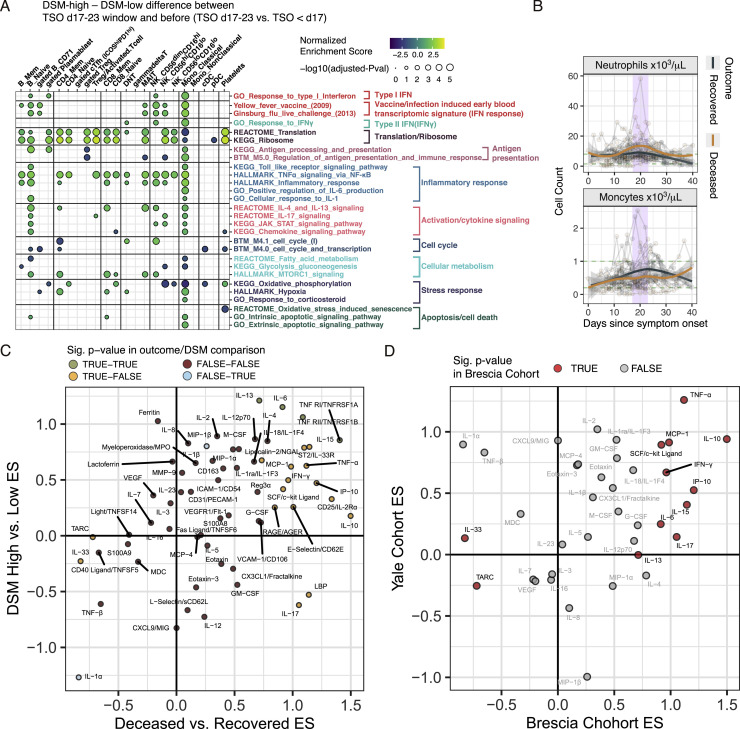
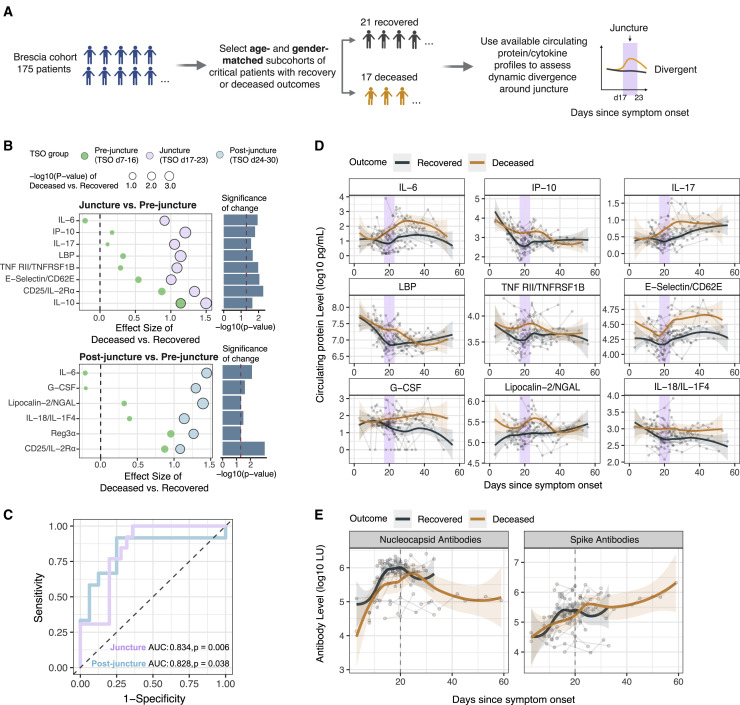
COVID-19 exhibits extensive patient-to-patient heterogeneity. To link immune response variation to disease severity and outcome over time, we longitudinally assessed circulating proteins as well as 188 surface protein markers, transcriptome, and T cell receptor sequence simultaneously in single peripheral immune cells from COVID-19 patients. Conditional-independence network analysis revealed primary correlates of disease severity, including gene expression signatures of apoptosis in plasmacytoid dendritic cells and attenuated inflammation but increased fatty acid metabolism in CD56dimCD16hi NK cells linked positively to circulating interleukin (IL)-15. CD8+ T cell activation was apparent without signs of exhaustion. Although cellular inflammation was depressed in severe patients early after hospitalization, it became elevated by days 17-23 post symptom onset, suggestive of a late wave of inflammatory responses. Furthermore, circulating protein trajectories at this time were divergent between and predictive of recovery versus fatal outcomes. Our findings stress the importance of timing in the analysis, clinical monitoring, and therapeutic intervention of COVID-19.
Keywords: CITE-seq; COVID-19; exhaustion; immune juncture; mixed-effect statistical modeling; single cell multi-omics; time-resolved.
Copyright © 2021. Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


Similar articles
-
Elevated Exhaustion Levels of NK and CD8+ T Cells as Indicators for Progression and Prognosis of COVID-19 Disease.Front Immunol. 2020 Oct 14;11:580237. doi: 10.3389/fimmu.2020.580237. eCollection 2020. Front Immunol. 2020. PMID: 33154753 Free PMC article.
-
Integrated analysis of circulating immune cellular and soluble mediators reveals specific COVID19 signatures at hospital admission with utility for prediction of clinical outcomes.Theranostics. 2022 Jan 1;12(1):290-306. doi: 10.7150/thno.63463. eCollection 2022. Theranostics. 2022. PMID: 34987646 Free PMC article.
-
NK and T Cell Immunological Signatures in Hospitalized Patients with COVID-19.Cells. 2021 Nov 15;10(11):3182. doi: 10.3390/cells10113182. Cells. 2021. PMID: 34831404 Free PMC article.
-
Exhausted NK cells and cytokine storms in COVID-19: Whether NK cell therapy could be a therapeutic choice.Hum Immunol. 2022 Jan;83(1):86-98. doi: 10.1016/j.humimm.2021.09.004. Epub 2021 Sep 8. Hum Immunol. 2022. PMID: 34583856 Free PMC article. Review.
-
T and NK Cell Phenotypic Abnormalities in Systemic Sclerosis: a Cohort Study and a Comprehensive Literature Review.Clin Rev Allergy Immunol. 2015 Dec;49(3):347-69. doi: 10.1007/s12016-015-8505-8. Clin Rev Allergy Immunol. 2015. PMID: 26445774 Review.
Cited by
-
A multi-organ map of the human immune system across age, sex and ethnicity.bioRxiv [Preprint]. 2024 Apr 29:2023.06.08.542671. doi: 10.1101/2023.06.08.542671. bioRxiv. 2024. PMID: 38746418 Free PMC article. Preprint.
-
Identifying Reproducible Transcription Regulator Coexpression Patterns with Single Cell Transcriptomics.bioRxiv [Preprint]. 2024 Oct 31:2024.02.15.580581. doi: 10.1101/2024.02.15.580581. bioRxiv. 2024. PMID: 38559016 Free PMC article. Preprint.
-
Gene expression profiling of host lipid metabolism in SARS-CoV-2 infected patients: a systematic review and integrated bioinformatics analysis.BMC Infect Dis. 2024 Jan 23;24(1):124. doi: 10.1186/s12879-024-08983-0. BMC Infect Dis. 2024. PMID: 38263024 Free PMC article.
-
Research and resource needs for understanding host immune responses to SARS-CoV-2 and COVID-19 vaccines during aging.Nat Aging. 2021 Dec;1(12):1073-1077. doi: 10.1038/s43587-021-00156-x. Epub 2021 Dec 22. Nat Aging. 2021. PMID: 36908301 Free PMC article.
-
Untimely TGFβ responses in COVID-19 limit antiviral functions of NK cells.Nature. 2021 Dec;600(7888):295-301. doi: 10.1038/s41586-021-04142-6. Epub 2021 Oct 25. Nature. 2021. PMID: 34695836
References
-
- Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.-H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., et al. HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. - PMC - PubMed
-
- Bates D., Mächler M., Bolker B., Walker S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015;67:1–48.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
