

A high-resolution protein architecture of the budding yeast genome
- PMID: 33692541
- PMCID: PMC8035251
- DOI: 10.1038/s41586-021-03314-8
A high-resolution protein architecture of the budding yeast genome
Abstract
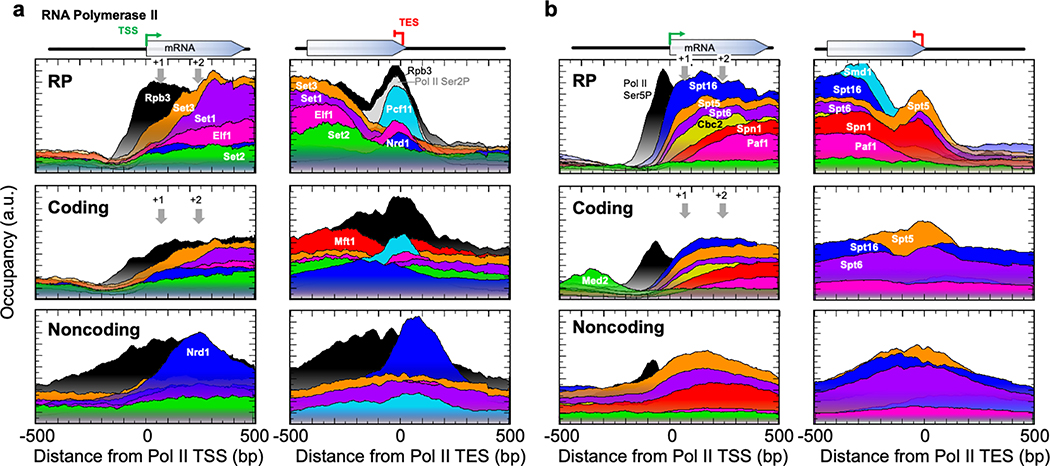
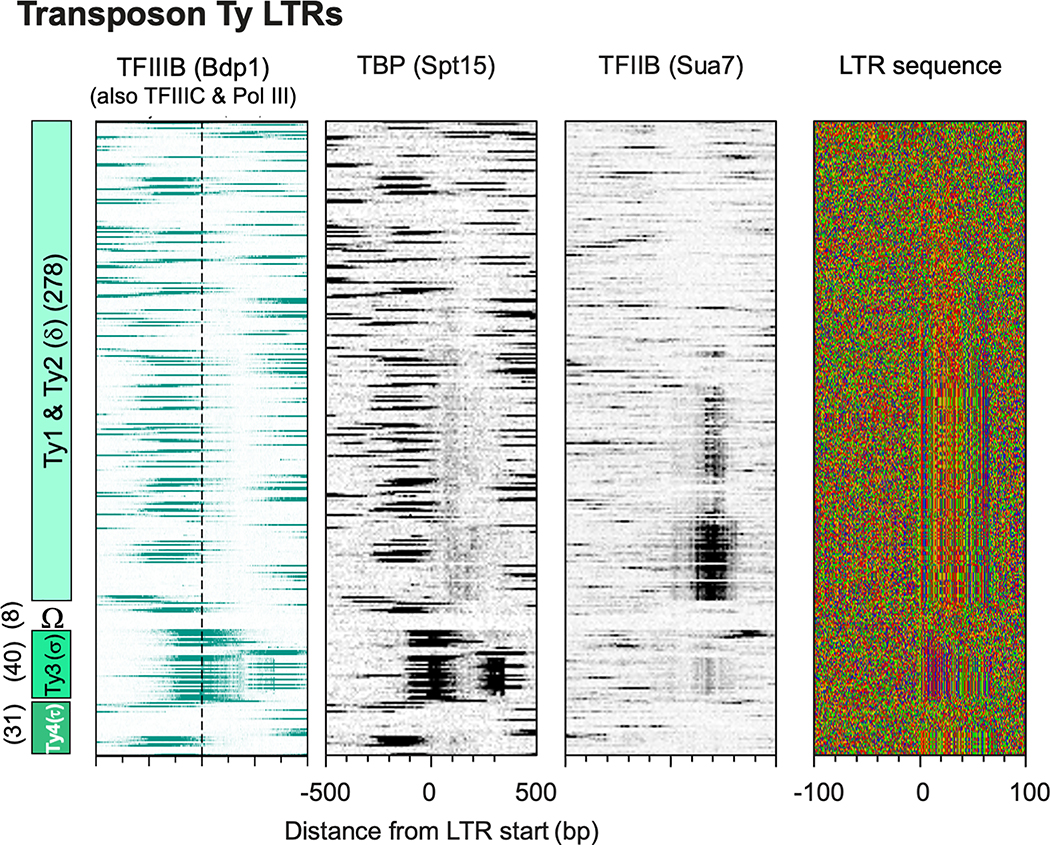
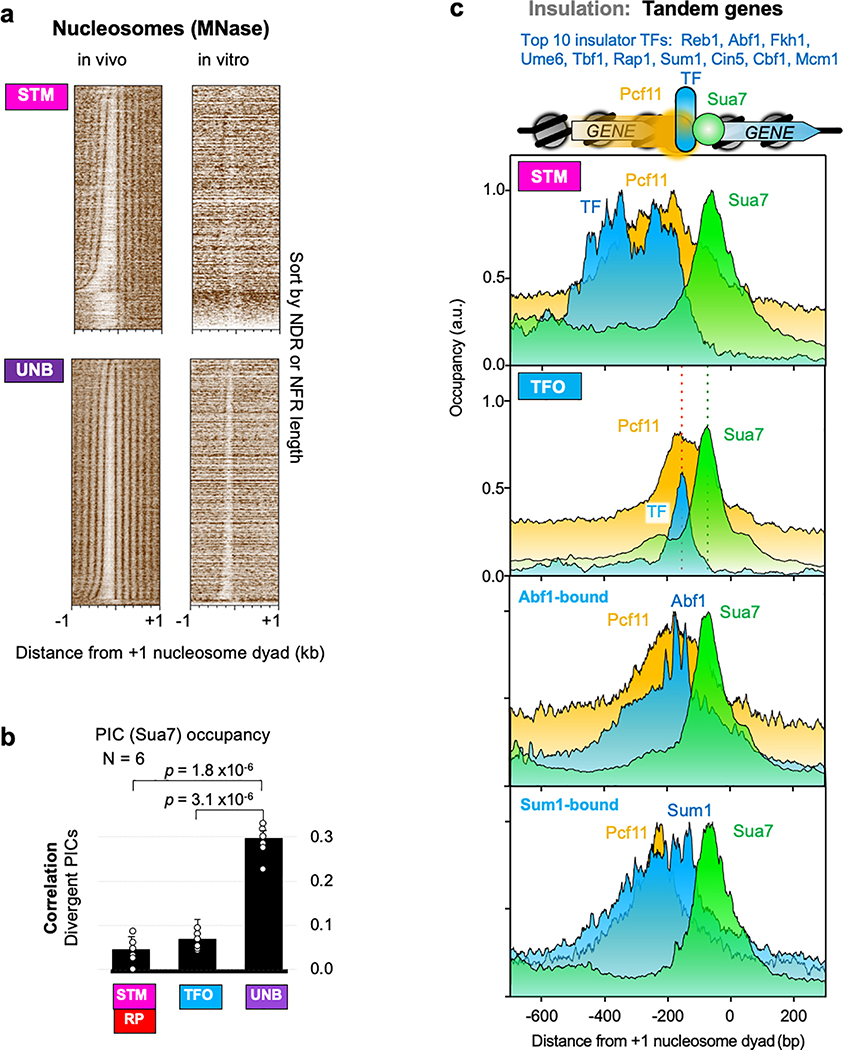
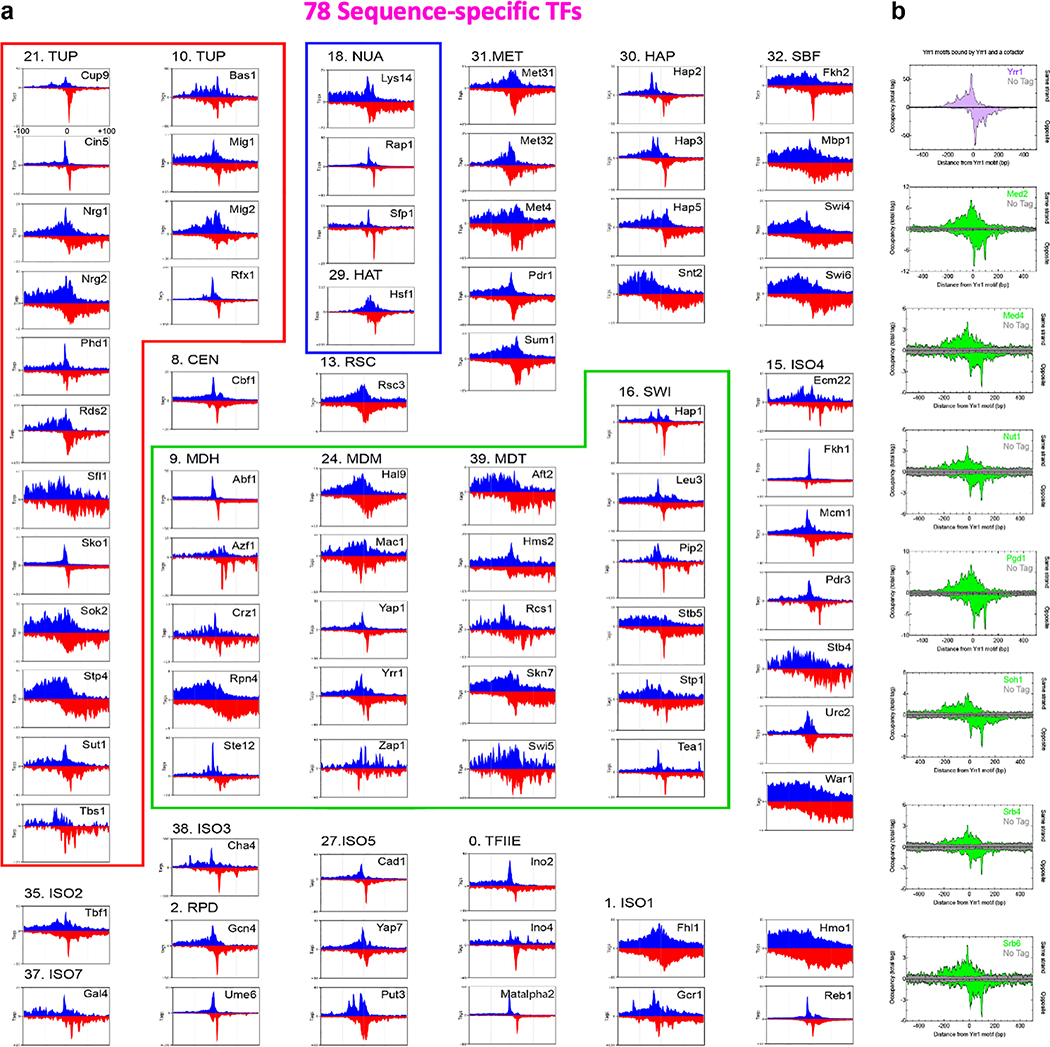
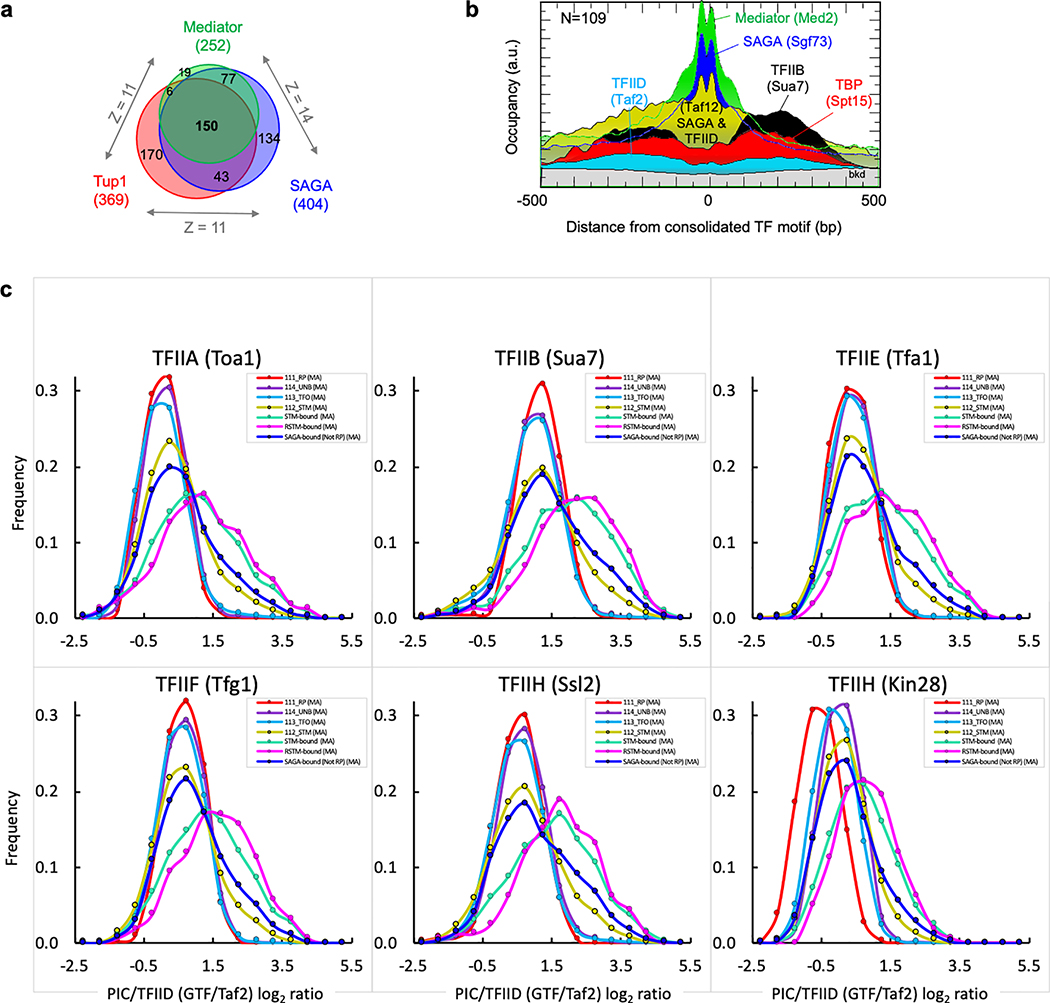
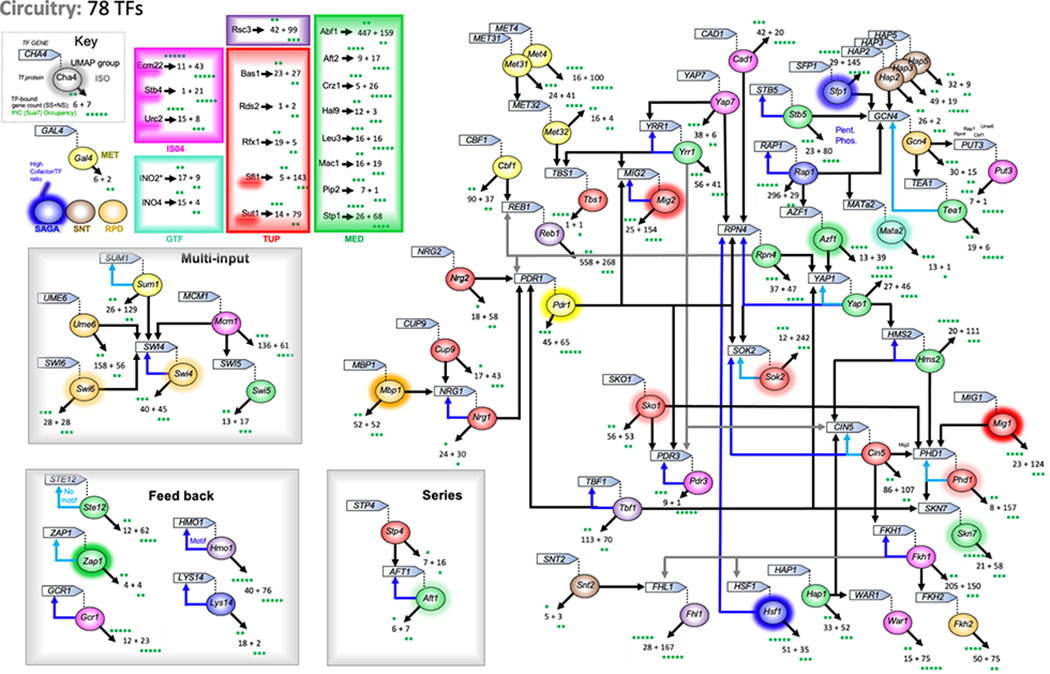
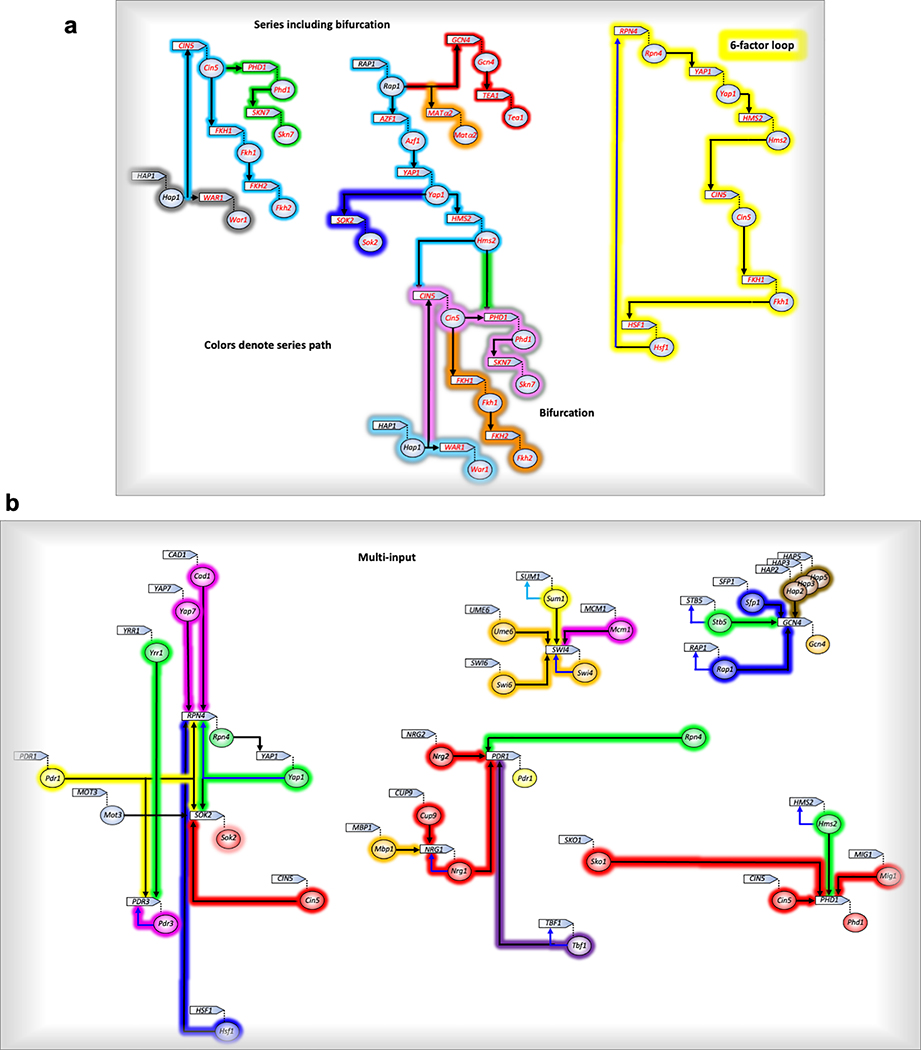
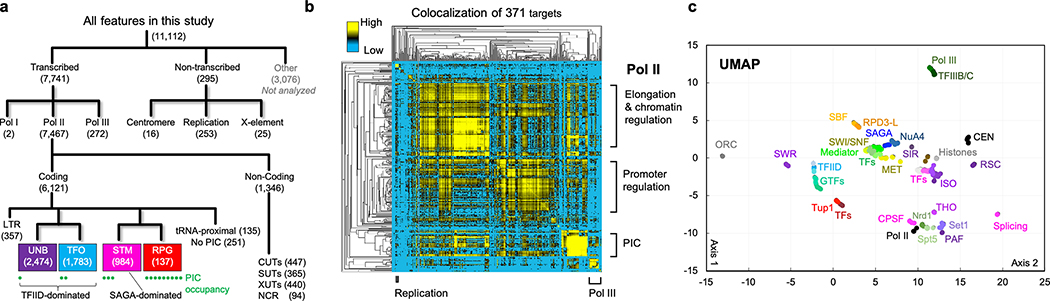
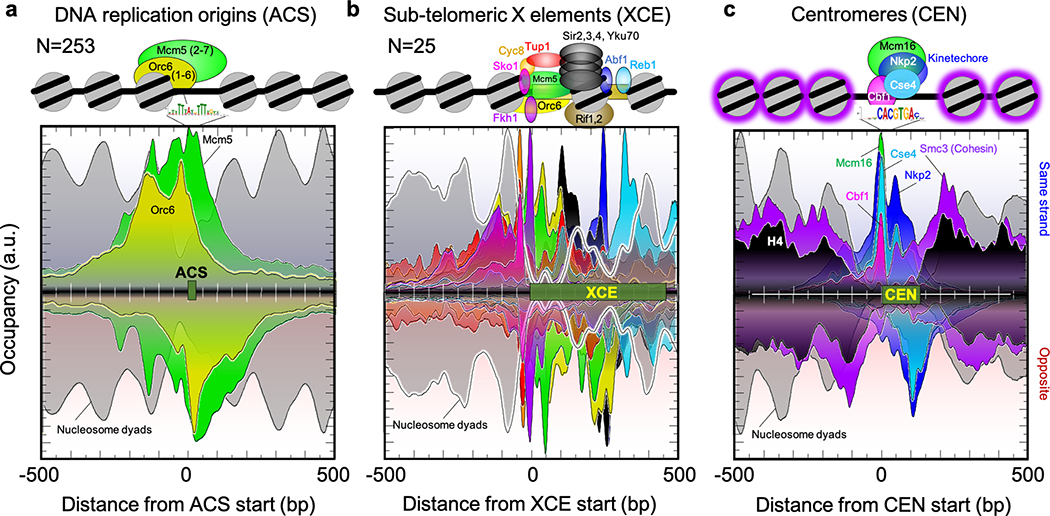
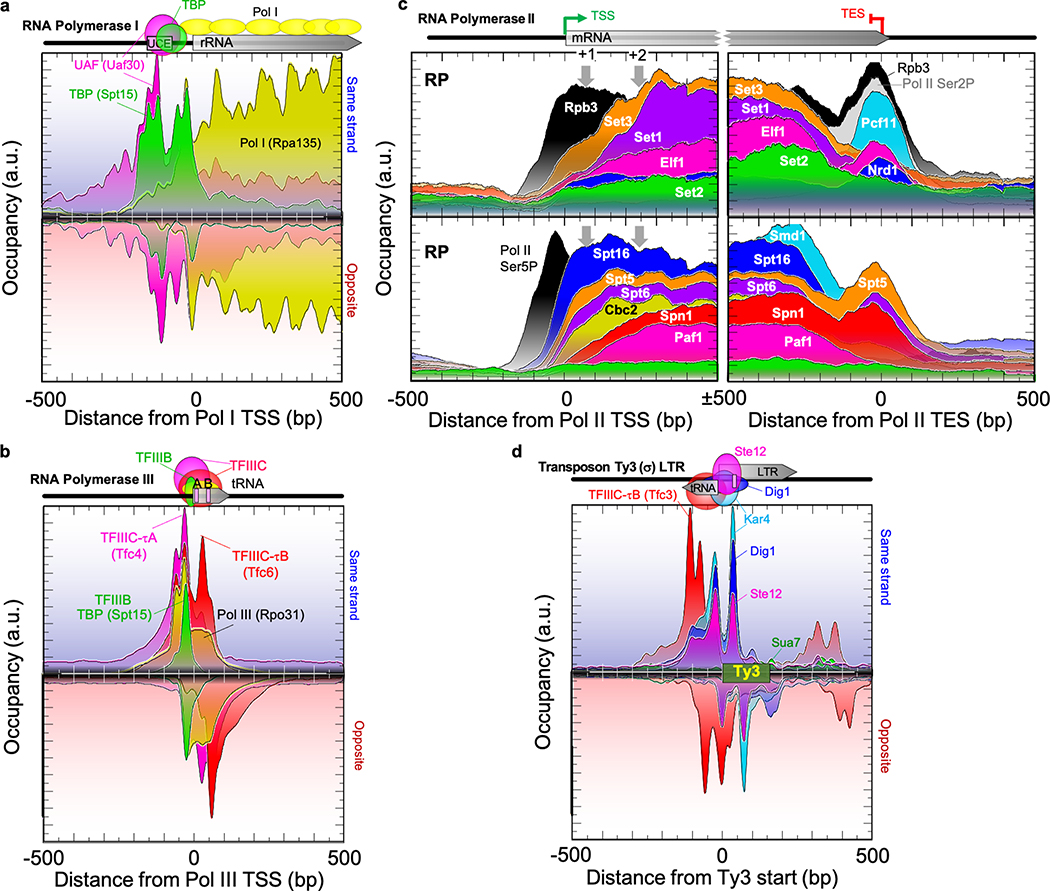
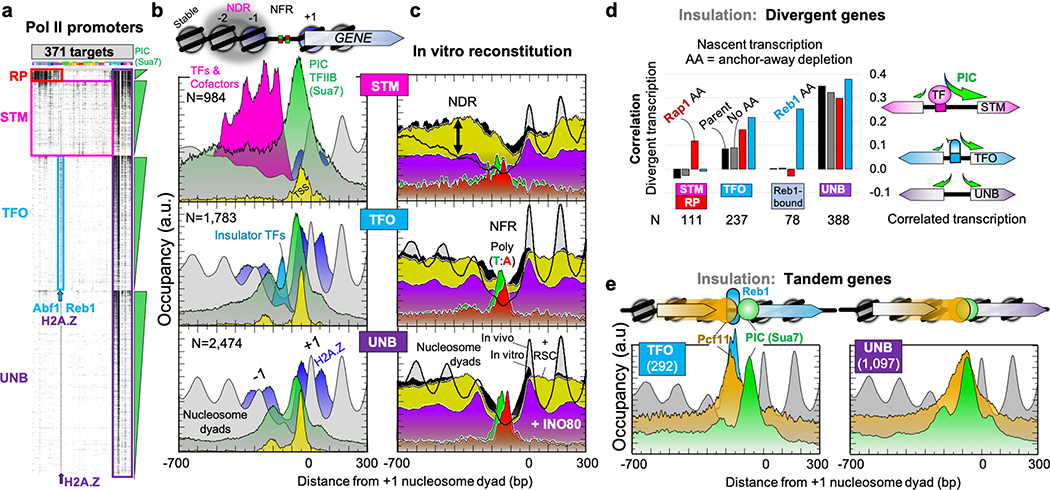
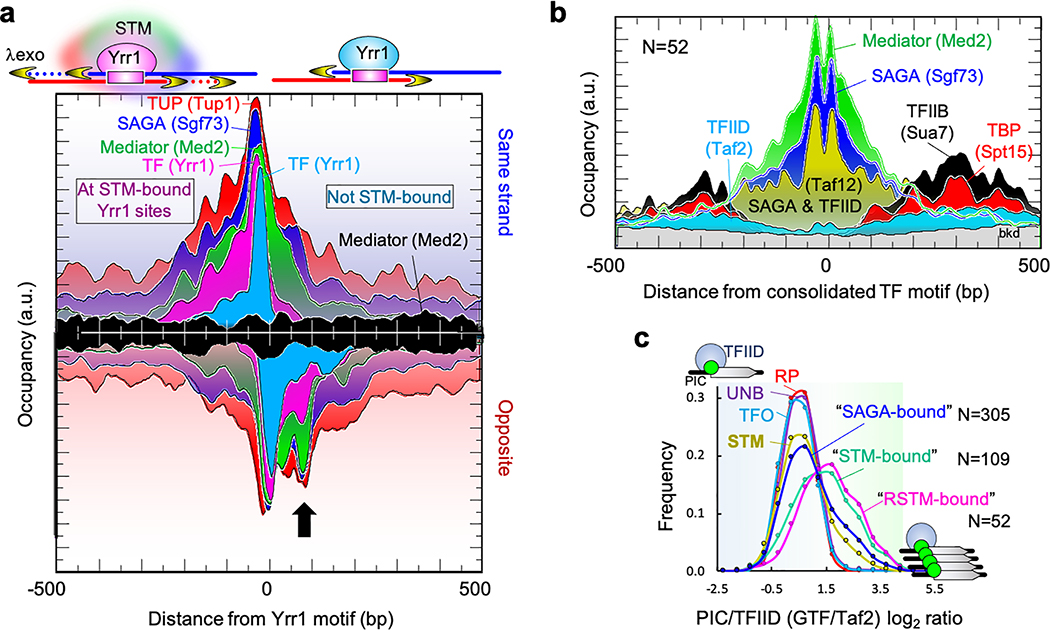
The genome-wide architecture of chromatin-associated proteins that maintains chromosome integrity and gene regulation is not well defined. Here we use chromatin immunoprecipitation, exonuclease digestion and DNA sequencing (ChIP-exo/seq)1,2 to define this architecture in Saccharomyces cerevisiae. We identify 21 meta-assemblages consisting of roughly 400 different proteins that are related to DNA replication, centromeres, subtelomeres, transposons and transcription by RNA polymerase (Pol) I, II and III. Replication proteins engulf a nucleosome, centromeres lack a nucleosome, and repressive proteins encompass three nucleosomes at subtelomeric X-elements. We find that most promoters associated with Pol II evolved to lack a regulatory region, having only a core promoter. These constitutive promoters comprise a short nucleosome-free region (NFR) adjacent to a +1 nucleosome, which together bind the transcription-initiation factor TFIID to form a preinitiation complex. Positioned insulators protect core promoters from upstream events. A small fraction of promoters evolved an architecture for inducibility, whereby sequence-specific transcription factors (ssTFs) create a nucleosome-depleted region (NDR) that is distinct from an NFR. We describe structural interactions among ssTFs, their cognate cofactors and the genome. These interactions include the nucleosomal and transcriptional regulators RPD3-L, SAGA, NuA4, Tup1, Mediator and SWI-SNF. Surprisingly, we do not detect interactions between ssTFs and TFIID, suggesting that such interactions do not stably occur. Our model for gene induction involves ssTFs, cofactors and general factors such as TBP and TFIIB, but not TFIID. By contrast, constitutive transcription involves TFIID but not ssTFs engaged with their cofactors. From this, we define a highly integrated network of gene regulation by ssTFs.
Conflict of interest statement
Figures
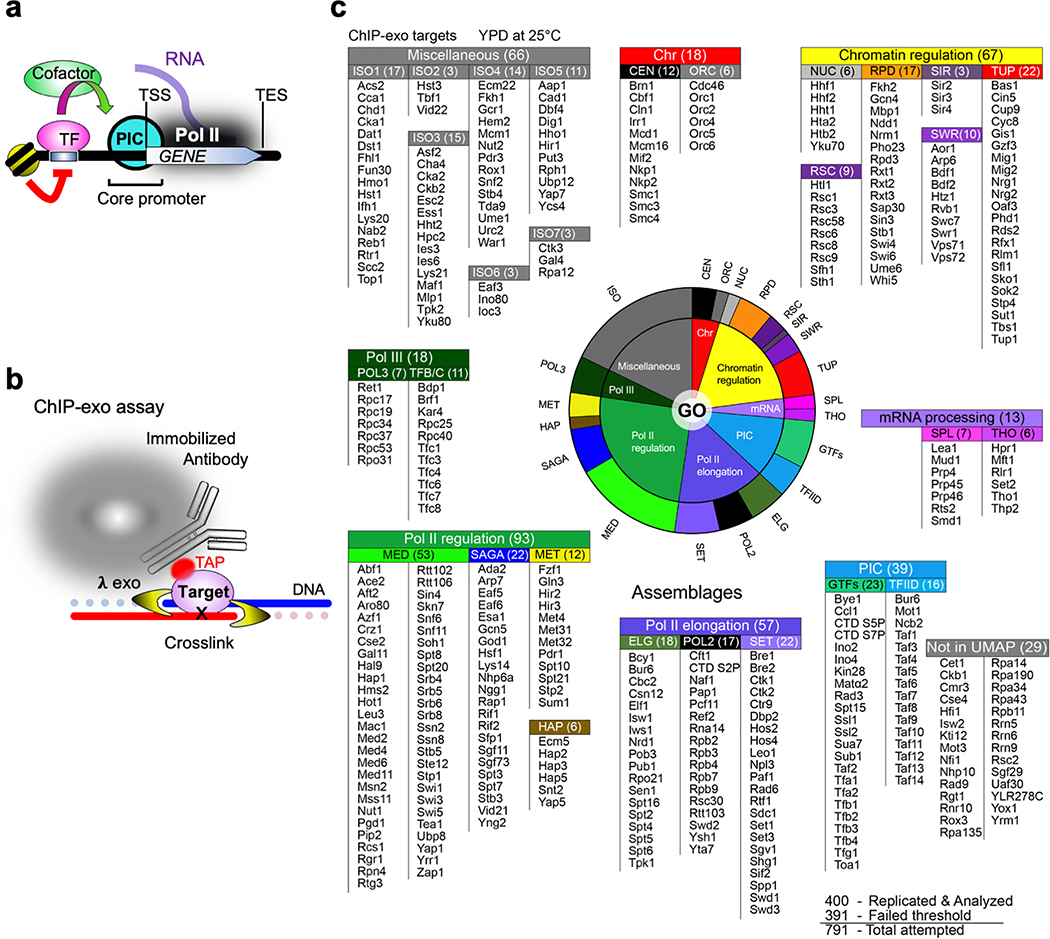
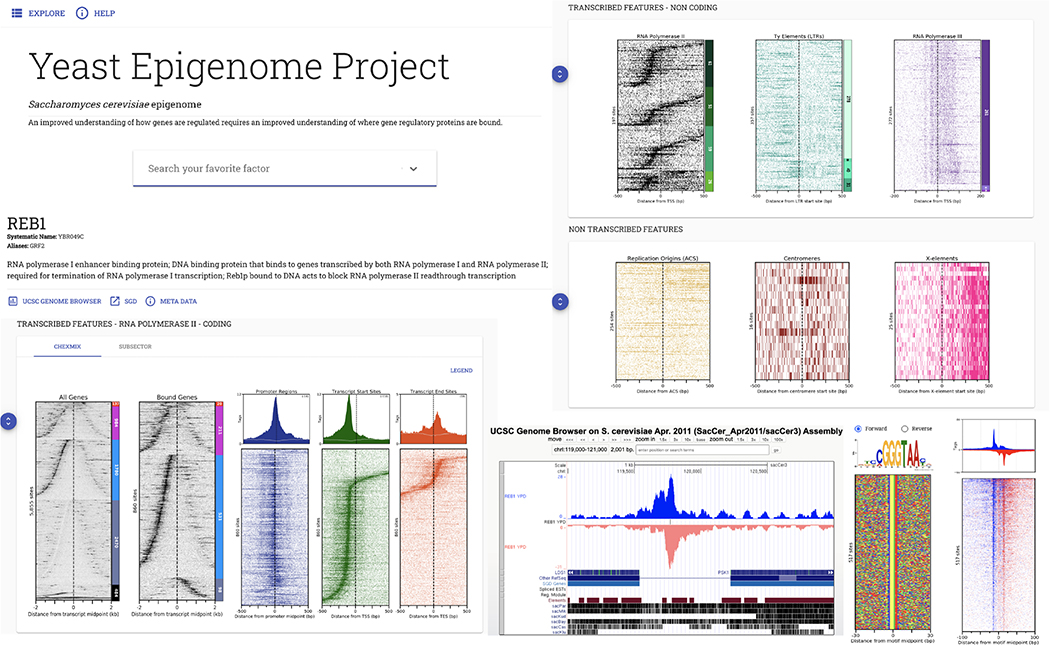
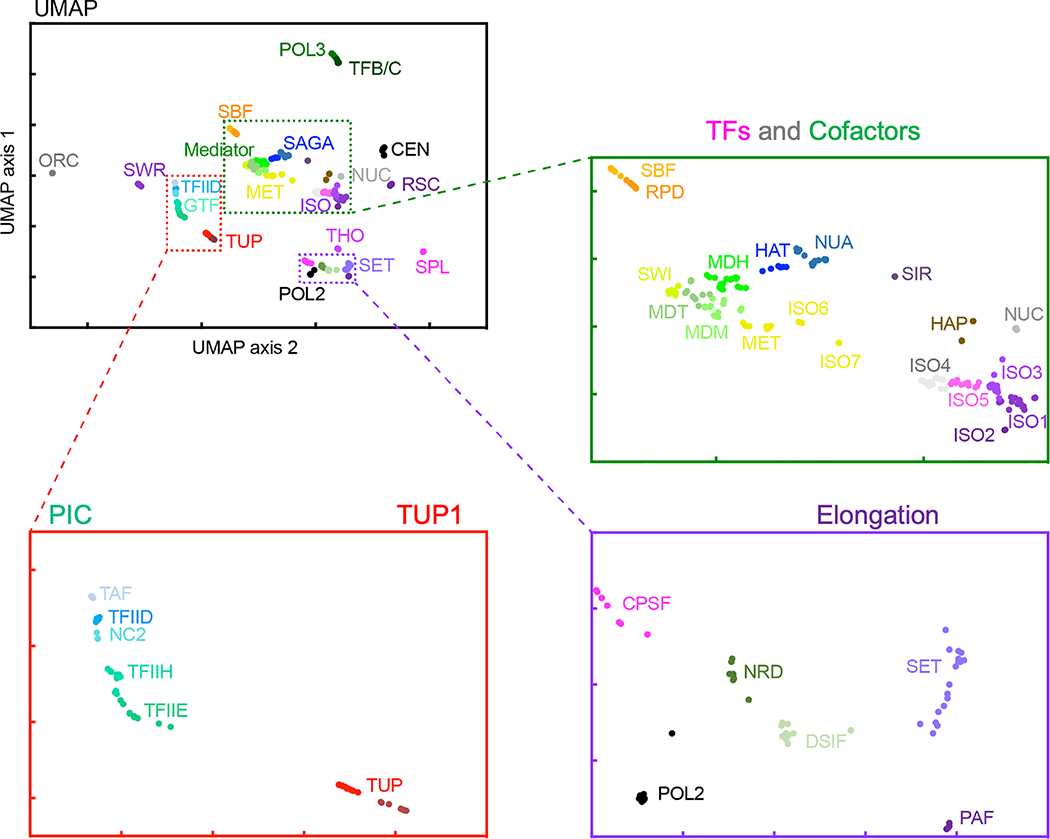
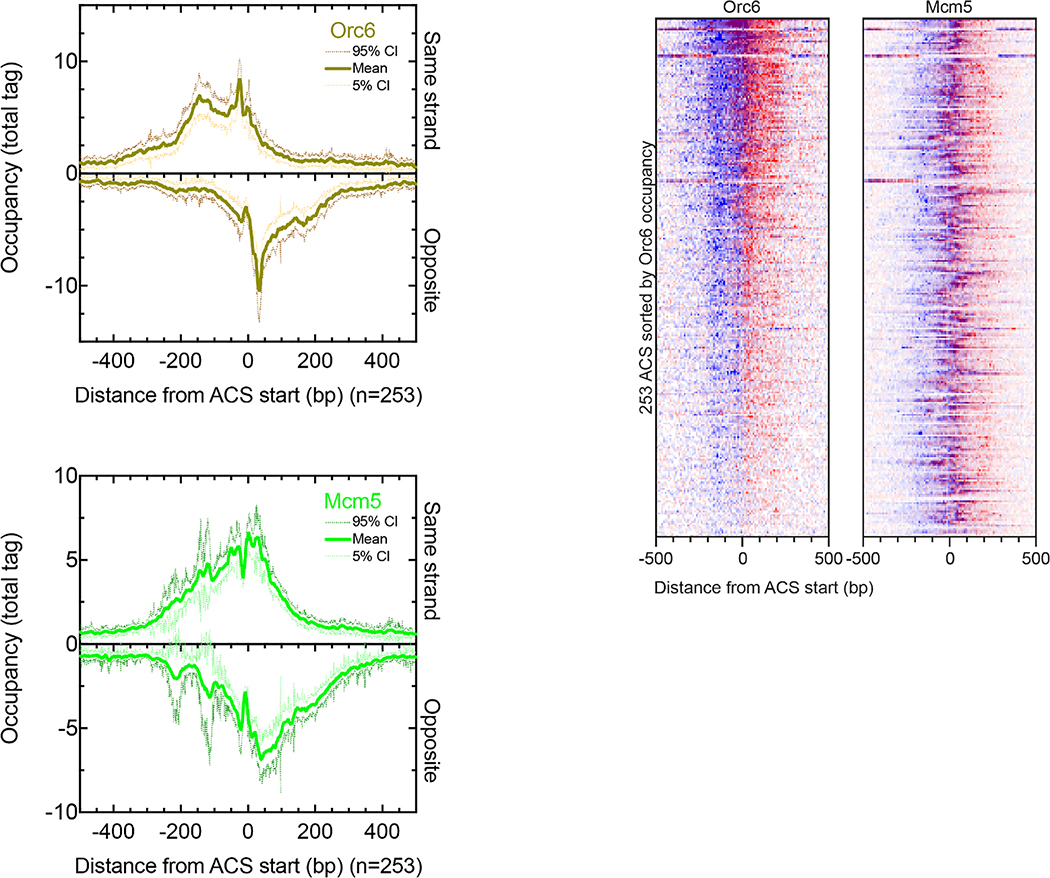
Similar articles
-
A canonical promoter organization of the transcription machinery and its regulators in the Saccharomyces genome.Genome Res. 2009 Mar;19(3):360-71. doi: 10.1101/gr.084970.108. Epub 2009 Jan 5. Genome Res. 2009. PMID: 19124666 Free PMC article.
-
Mediator, TATA-binding protein, and RNA polymerase II contribute to low histone occupancy at active gene promoters in yeast.J Biol Chem. 2014 May 23;289(21):14981-95. doi: 10.1074/jbc.M113.529354. Epub 2014 Apr 11. J Biol Chem. 2014. PMID: 24727477 Free PMC article.
-
Basal core promoters control the equilibrium between negative cofactor 2 and preinitiation complexes in human cells.Genome Biol. 2010;11(3):R33. doi: 10.1186/gb-2010-11-3-r33. Epub 2010 Mar 15. Genome Biol. 2010. PMID: 20230619 Free PMC article.
-
Architecture of the multi-functional SAGA complex and the molecular mechanism of holding TBP.FEBS J. 2021 May;288(10):3135-3147. doi: 10.1111/febs.15563. Epub 2020 Sep 29. FEBS J. 2021. PMID: 32946670 Review.
-
The general transcription machinery and general cofactors.Crit Rev Biochem Mol Biol. 2006 May-Jun;41(3):105-78. doi: 10.1080/10409230600648736. Crit Rev Biochem Mol Biol. 2006. PMID: 16858867 Review.
Cited by
-
TFIIB-Termination Factor Interaction Affects Termination of Transcription on Genome-Wide Scale.Int J Mol Sci. 2024 Aug 8;25(16):8643. doi: 10.3390/ijms25168643. Int J Mol Sci. 2024. PMID: 39201330 Free PMC article.
-
Systematic dissection of sequence features affecting binding specificity of a pioneer factor reveals binding synergy between FOXA1 and AP-1.Mol Cell. 2024 Aug 8;84(15):2838-2855.e10. doi: 10.1016/j.molcel.2024.06.022. Epub 2024 Jul 16. Mol Cell. 2024. PMID: 39019045
-
In vitro reconstitution of chromatin domains shows a role for nucleosome positioning in 3D genome organization.Nat Genet. 2024 Mar;56(3):483-492. doi: 10.1038/s41588-023-01649-8. Epub 2024 Jan 30. Nat Genet. 2024. PMID: 38291333 Free PMC article.
-
An integrated SAGA and TFIID PIC assembly pathway selective for poised and induced promoters.Genes Dev. 2022 Sep 1;36(17-18):985-1001. doi: 10.1101/gad.350026.122. Epub 2022 Oct 27. Genes Dev. 2022. PMID: 36302553 Free PMC article.
-
High-Resolution Deep Sequencing of Nascent Transcription in Yeast with BioGRO-seq.Methods Mol Biol. 2022;2477:57-70. doi: 10.1007/978-1-0716-2257-5_4. Methods Mol Biol. 2022. PMID: 35524111
References
-
- Cramer P Organization and regulation of gene transcription. Nature 573, 45–54 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
