

Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family
- PMID: 33536238
- PMCID: PMC7898563
- DOI: 10.26508/lsa.202000858
Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family
Abstract
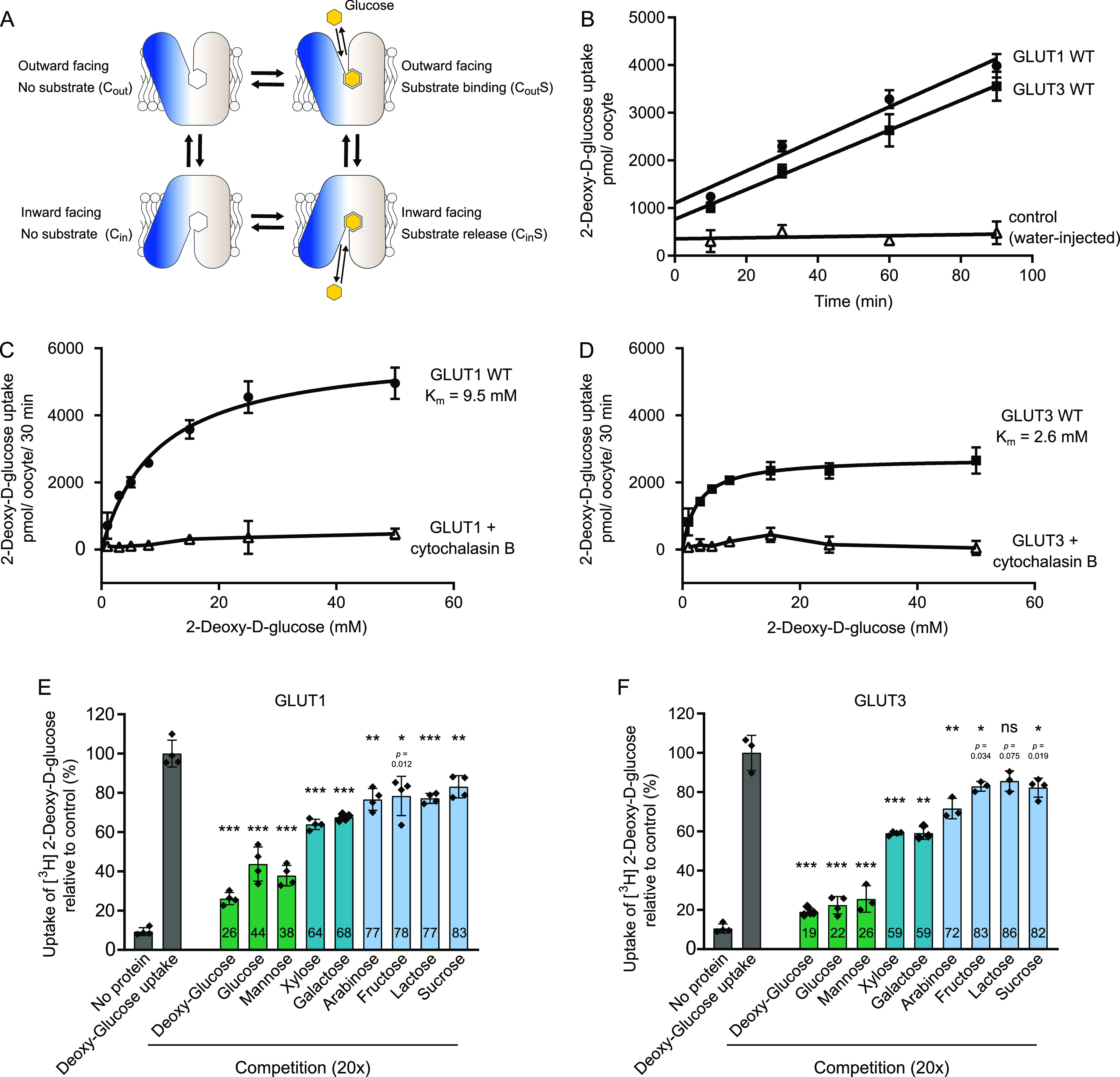
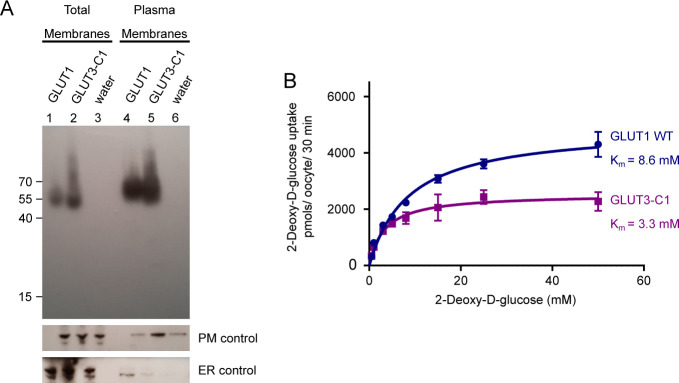
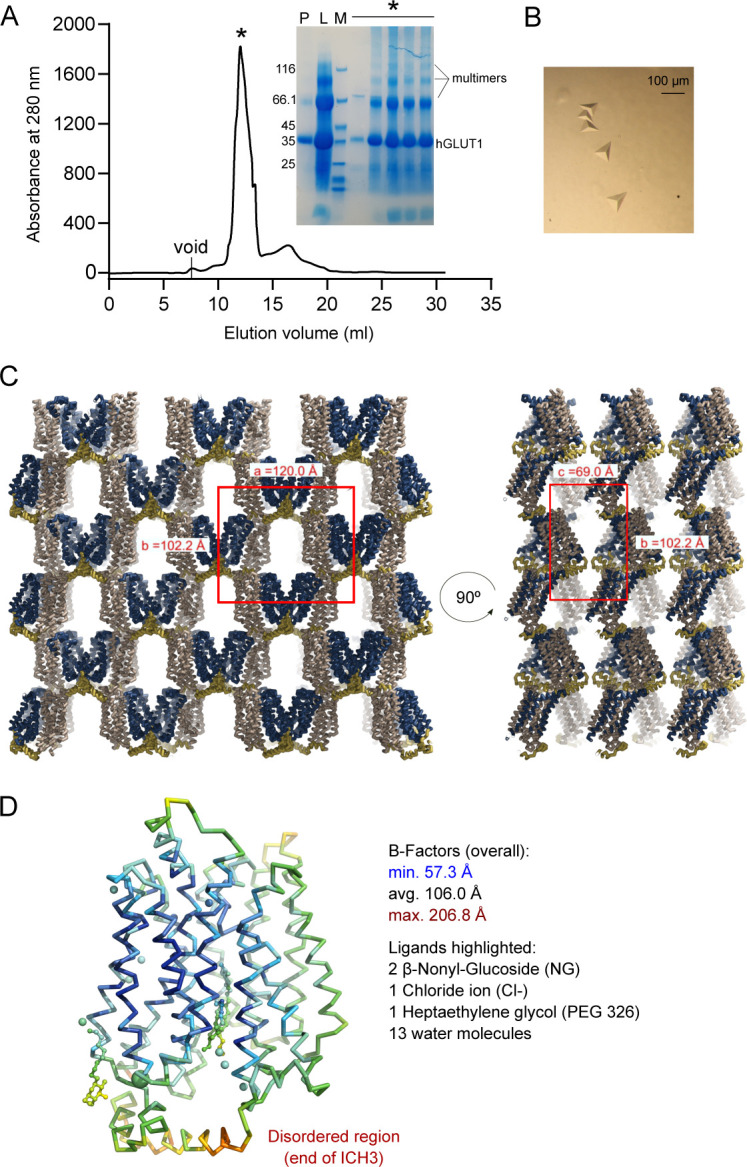
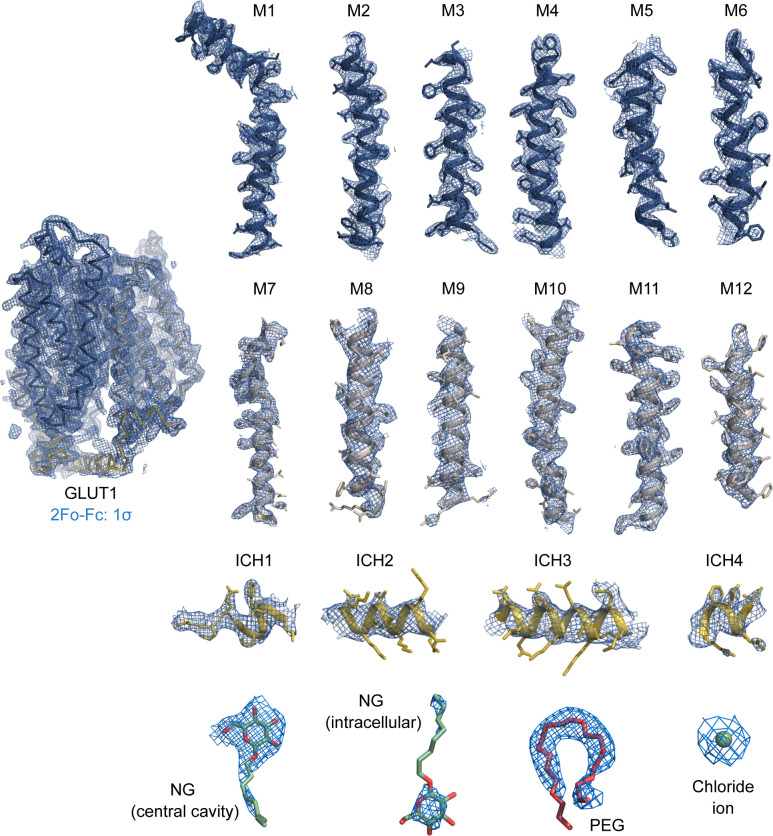
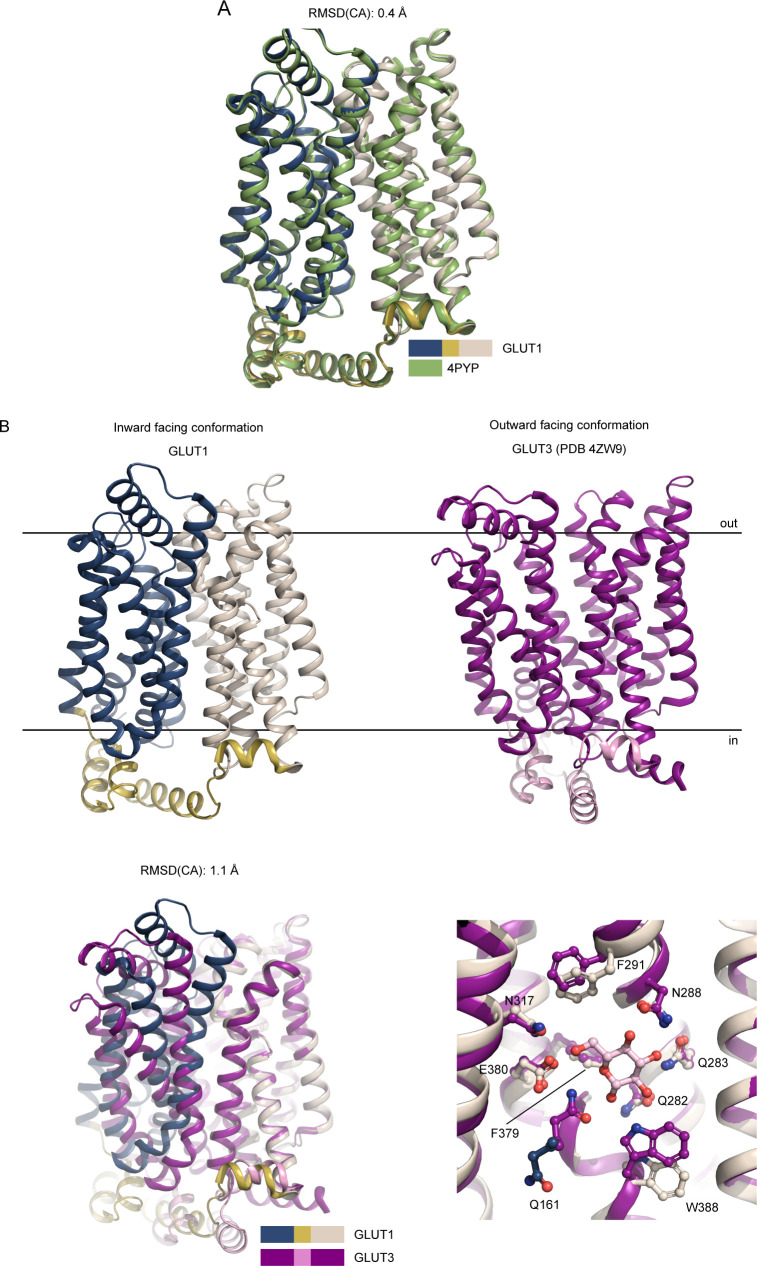
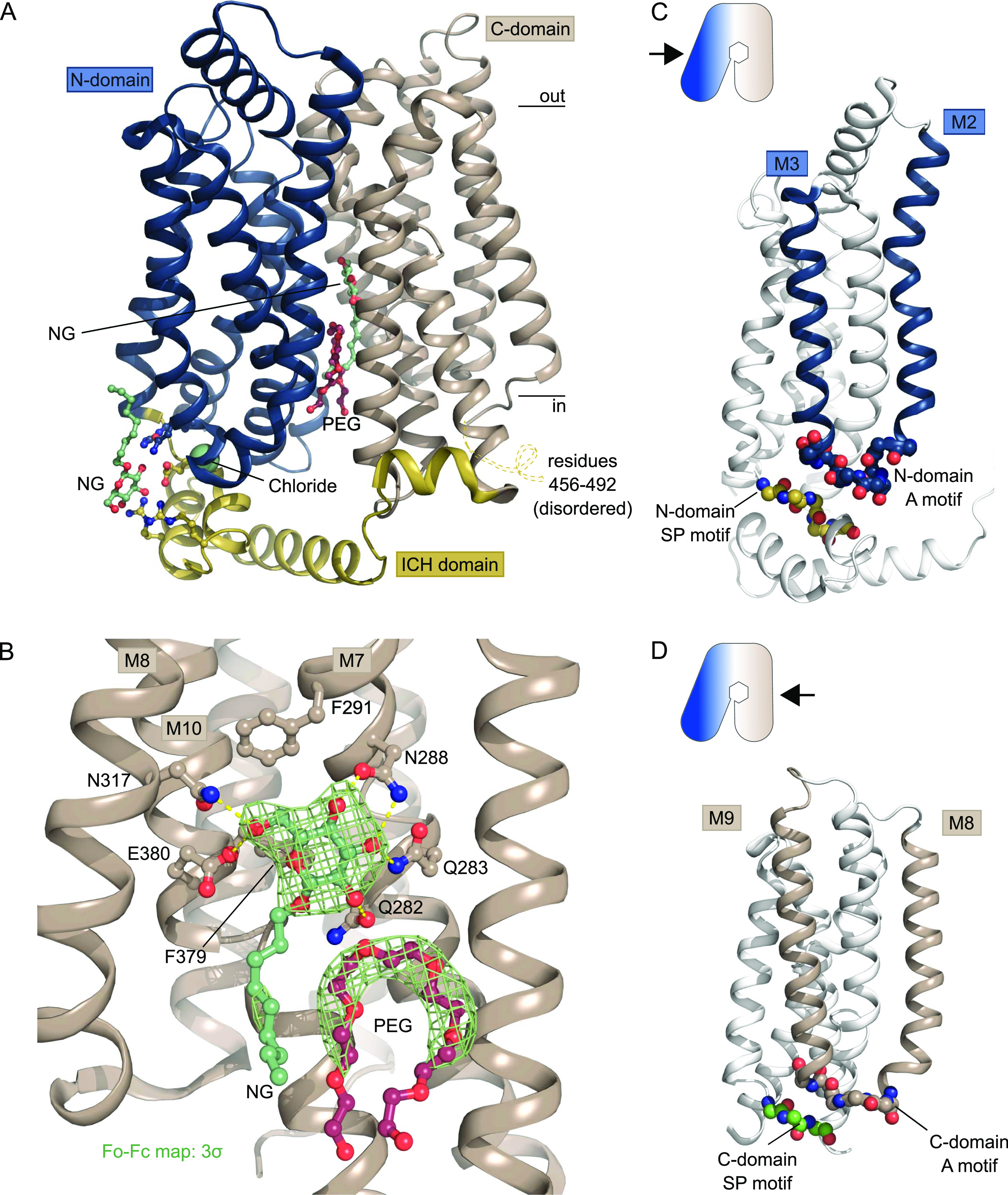
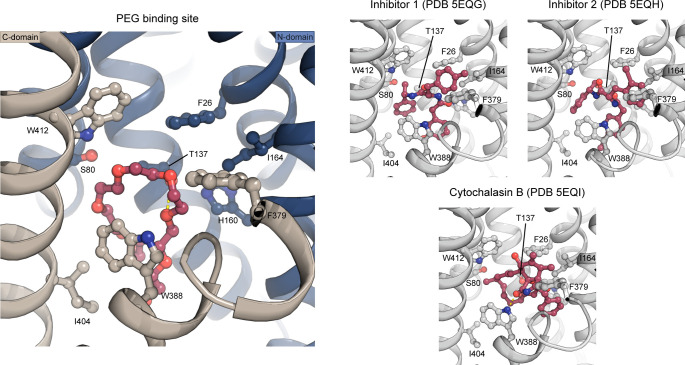
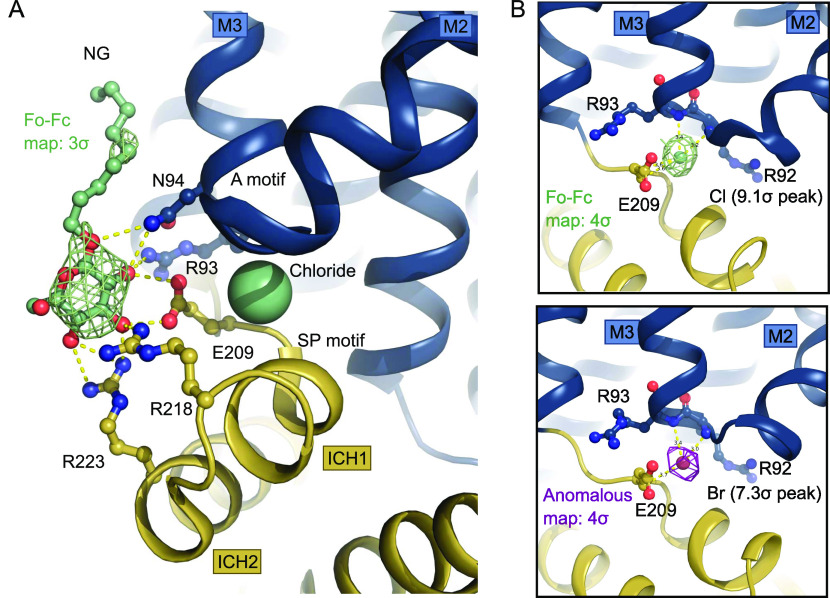
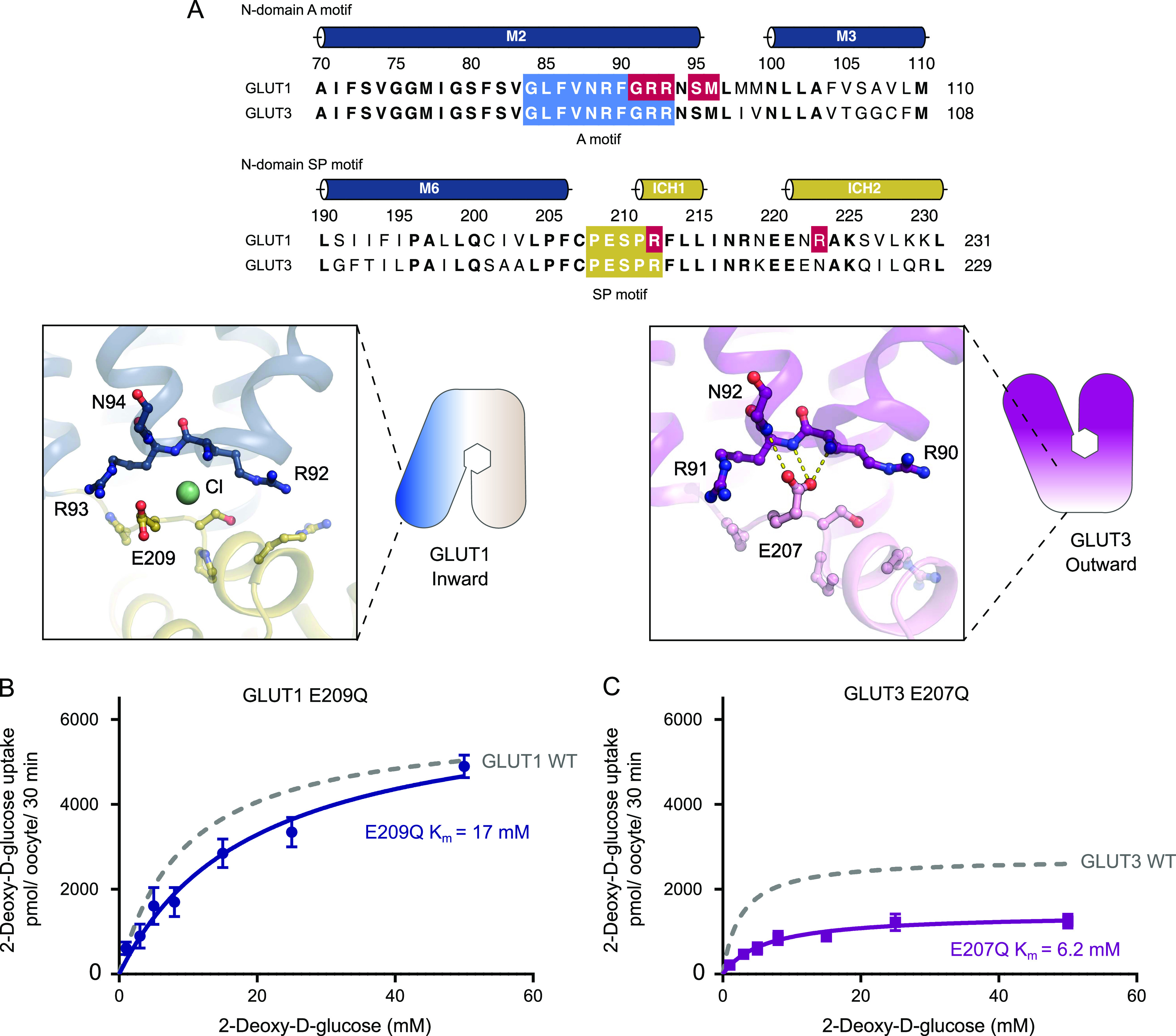
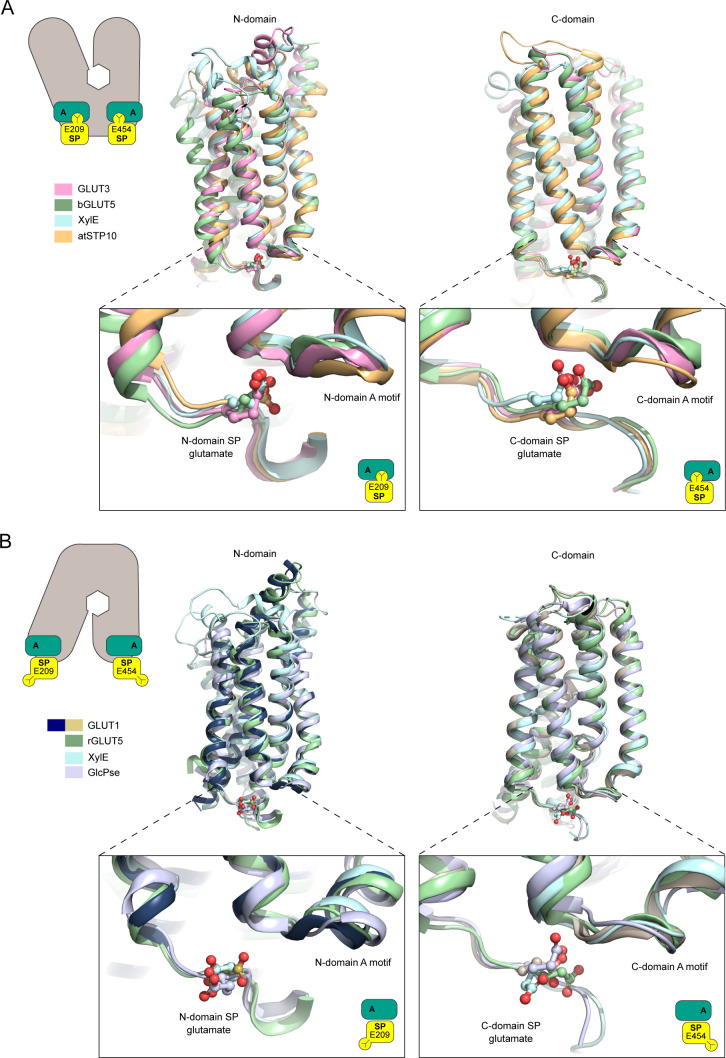
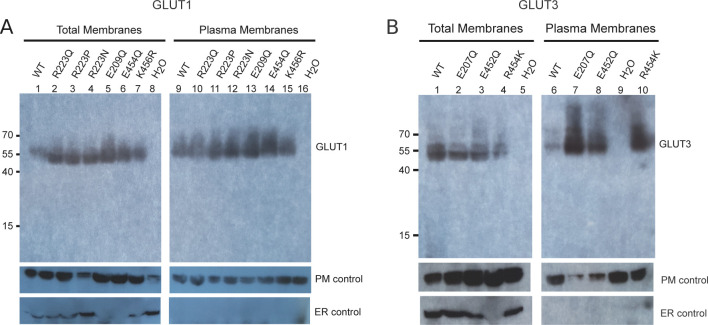
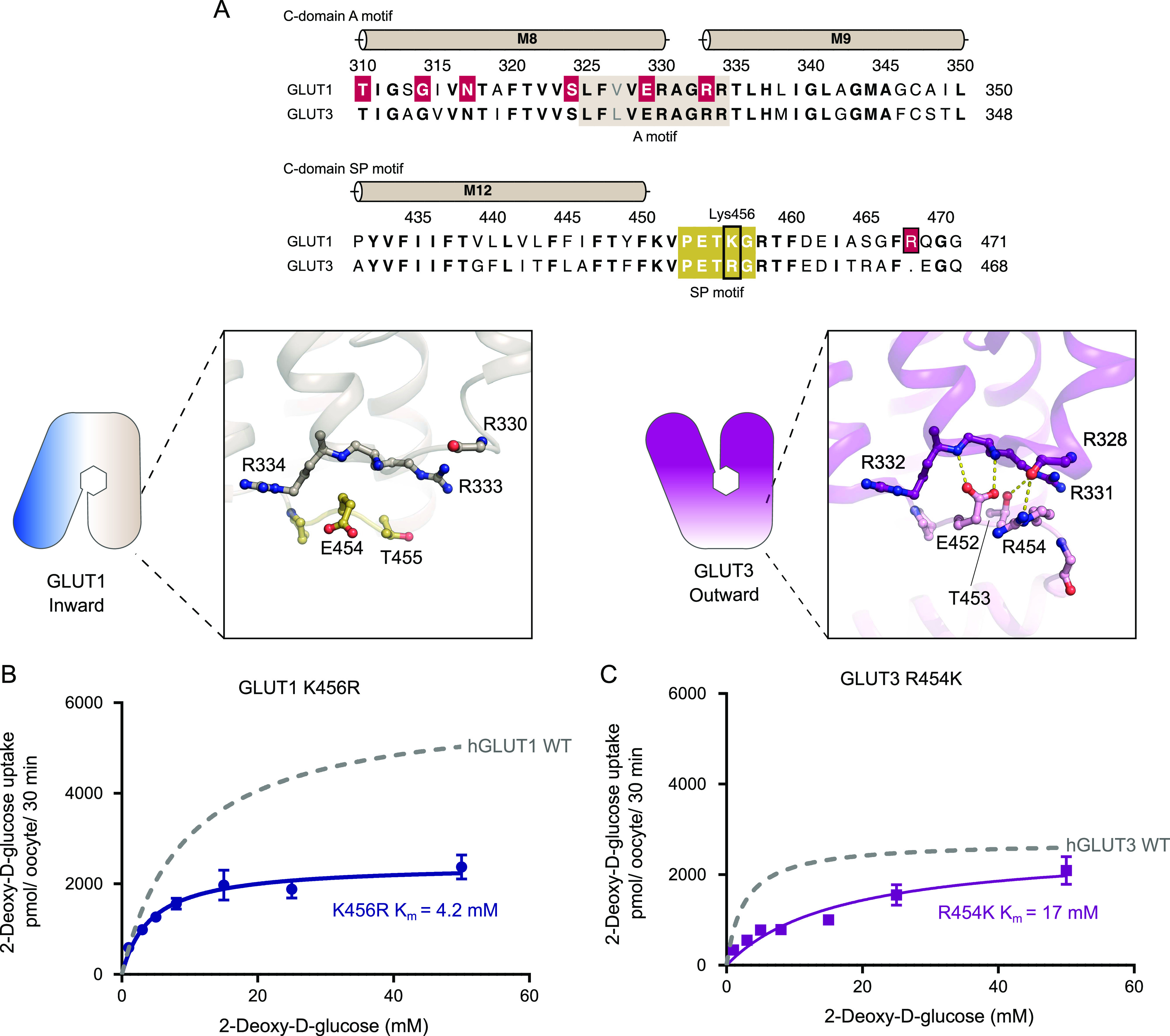
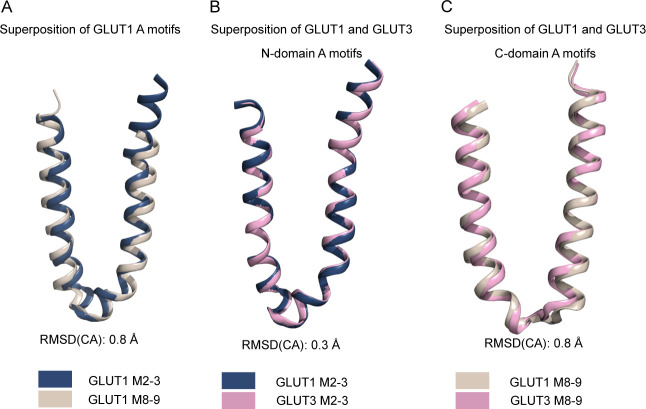
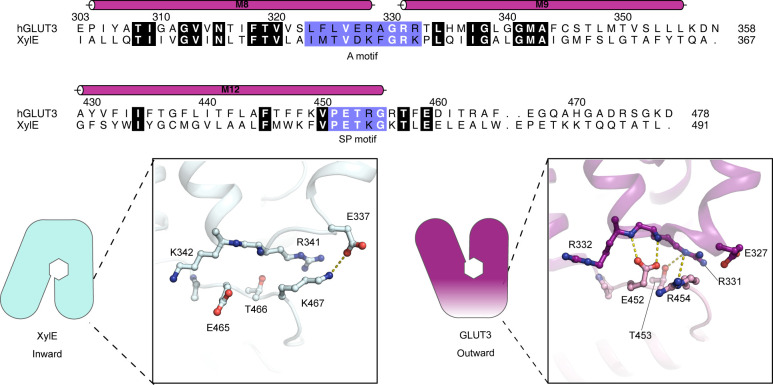
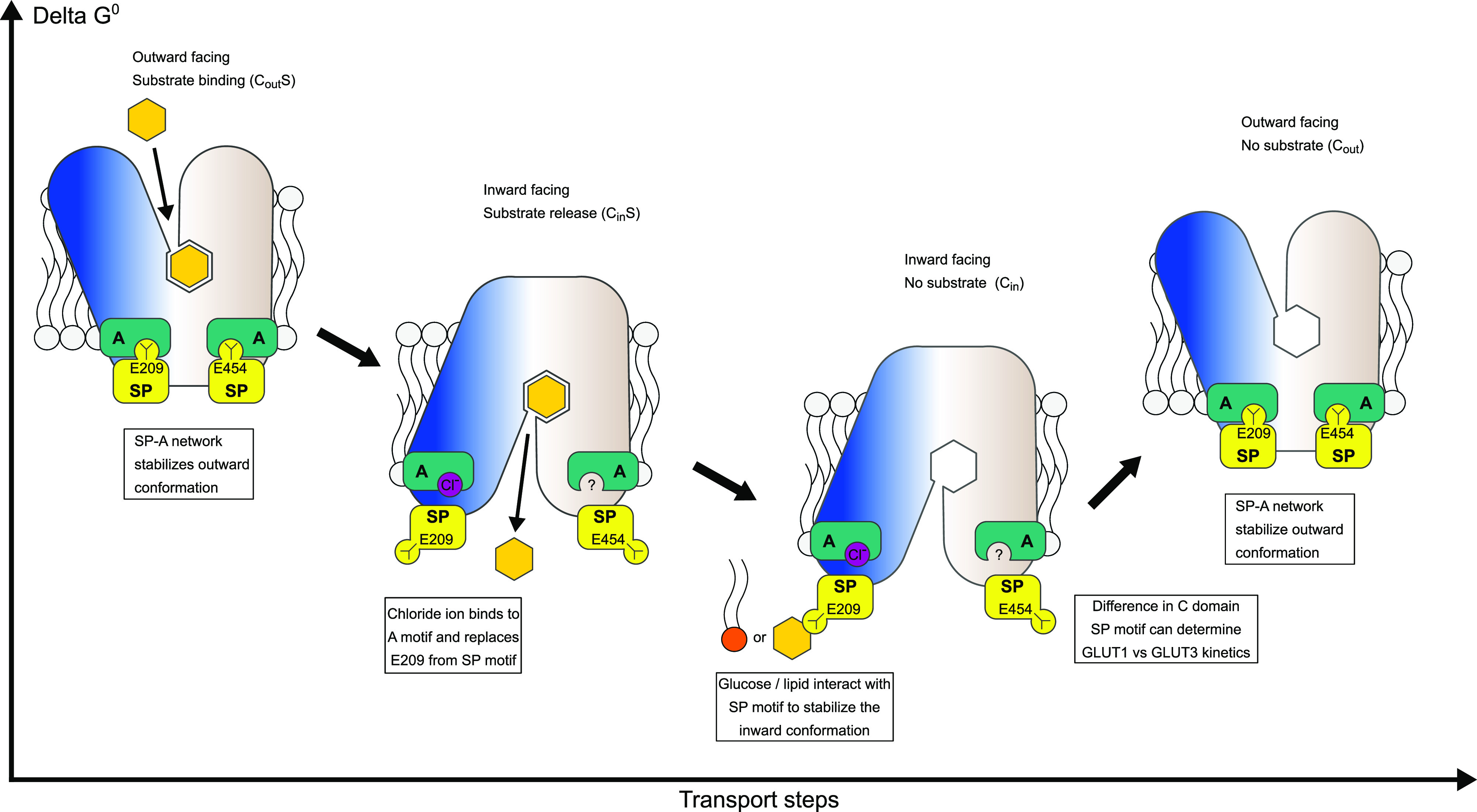
The human glucose transporters GLUT1 and GLUT3 have a central role in glucose uptake as canonical members of the Sugar Porter (SP) family. GLUT1 and GLUT3 share a fully conserved substrate-binding site with identical substrate coordination, but differ significantly in transport affinity in line with their physiological function. Here, we present a 2.4 Å crystal structure of GLUT1 in an inward open conformation and compare it with GLUT3 using both structural and functional data. Our work shows that interactions between a cytosolic "SP motif" and a conserved "A motif" stabilize the outward conformational state and increases substrate apparent affinity. Furthermore, we identify a previously undescribed Cl- ion site in GLUT1 and an endofacial lipid/glucose binding site which modulate GLUT kinetics. The results provide a possible explanation for the difference between GLUT1 and GLUT3 glucose affinity, imply a general model for the kinetic regulation in GLUTs and suggest a physiological function for the defining SP sequence motif in the SP family.
© 2021 Custódio et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Similar articles
-
Crystallization and Structural Determination of the Human Glucose Transporters GLUT1 and GLUT3.Methods Mol Biol. 2018;1713:15-29. doi: 10.1007/978-1-4939-7507-5_2. Methods Mol Biol. 2018. PMID: 29218514
-
Reconciling contradictory findings: Glucose transporter 1 (GLUT1) functions as an oligomer of allosteric, alternating access transporters.J Biol Chem. 2017 Dec 22;292(51):21035-21046. doi: 10.1074/jbc.M117.815589. Epub 2017 Oct 24. J Biol Chem. 2017. PMID: 29066623 Free PMC article.
-
Molecular basis of ligand recognition and transport by glucose transporters.Nature. 2015 Oct 15;526(7573):391-6. doi: 10.1038/nature14655. Epub 2015 Jul 15. Nature. 2015. PMID: 26176916
-
Pathogenic mutations causing glucose transport defects in GLUT1 transporter: The role of intermolecular forces in protein structure-function.Biophys Chem. 2015 May-Jun;200-201:9-17. doi: 10.1016/j.bpc.2015.03.005. Epub 2015 Mar 25. Biophys Chem. 2015. PMID: 25863194 Review.
-
Will the original glucose transporter isoform please stand up!Am J Physiol Endocrinol Metab. 2009 Oct;297(4):E836-48. doi: 10.1152/ajpendo.00496.2009. Epub 2009 Aug 18. Am J Physiol Endocrinol Metab. 2009. PMID: 19690067 Free PMC article. Review.
Cited by
-
Multiple roles for the cytoplasmic C-terminal domains of the yeast cell surface receptors Rgt2 and Snf3 in glucose sensing and signaling.Sci Rep. 2024 Feb 19;14(1):4055. doi: 10.1038/s41598-024-54628-2. Sci Rep. 2024. PMID: 38374219 Free PMC article.
-
Structural basis of promiscuous substrate transport by Organic Cation Transporter 1.Nat Commun. 2023 Oct 11;14(1):6374. doi: 10.1038/s41467-023-42086-9. Nat Commun. 2023. PMID: 37821493 Free PMC article.
-
Identification of Structural Determinants of the Transport of the Dehydroascorbic Acid Mediated by Glucose Transport GLUT1.Molecules. 2023 Jan 5;28(2):521. doi: 10.3390/molecules28020521. Molecules. 2023. PMID: 36677580 Free PMC article.
-
The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming.Cell Metab. 2022 Apr 5;34(4):516-532.e11. doi: 10.1016/j.cmet.2022.02.015. Epub 2022 Mar 21. Cell Metab. 2022. PMID: 35316657 Free PMC article.
-
An appraisal of the current status of inhibition of glucose transporters as an emerging antineoplastic approach: Promising potential of new pan-GLUT inhibitors.Front Pharmacol. 2022 Nov 1;13:1035510. doi: 10.3389/fphar.2022.1035510. eCollection 2022. Front Pharmacol. 2022. PMID: 36386187 Free PMC article. Review.
References
-
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. 10.1107/s0907444909052925 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous