

Neuronal hyperexcitability is a DLK-dependent trigger of herpes simplex virus reactivation that can be induced by IL-1
- PMID: 33350386
- PMCID: PMC7773336
- DOI: 10.7554/eLife.58037
Neuronal hyperexcitability is a DLK-dependent trigger of herpes simplex virus reactivation that can be induced by IL-1
Abstract
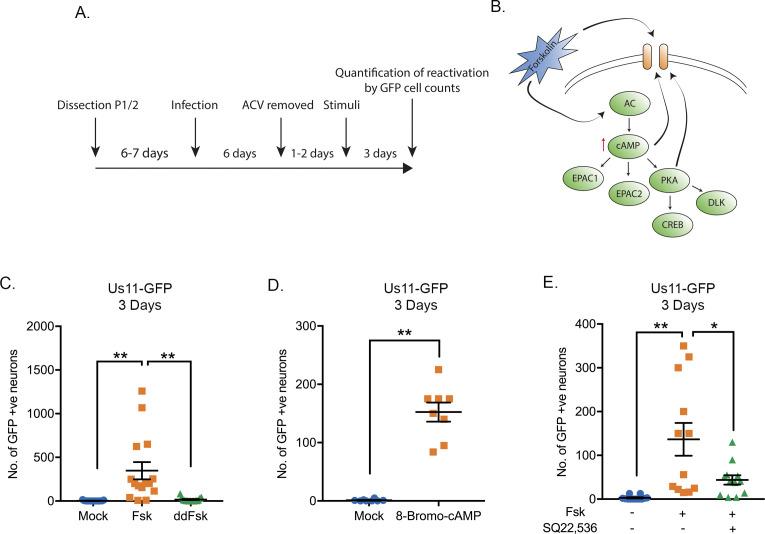
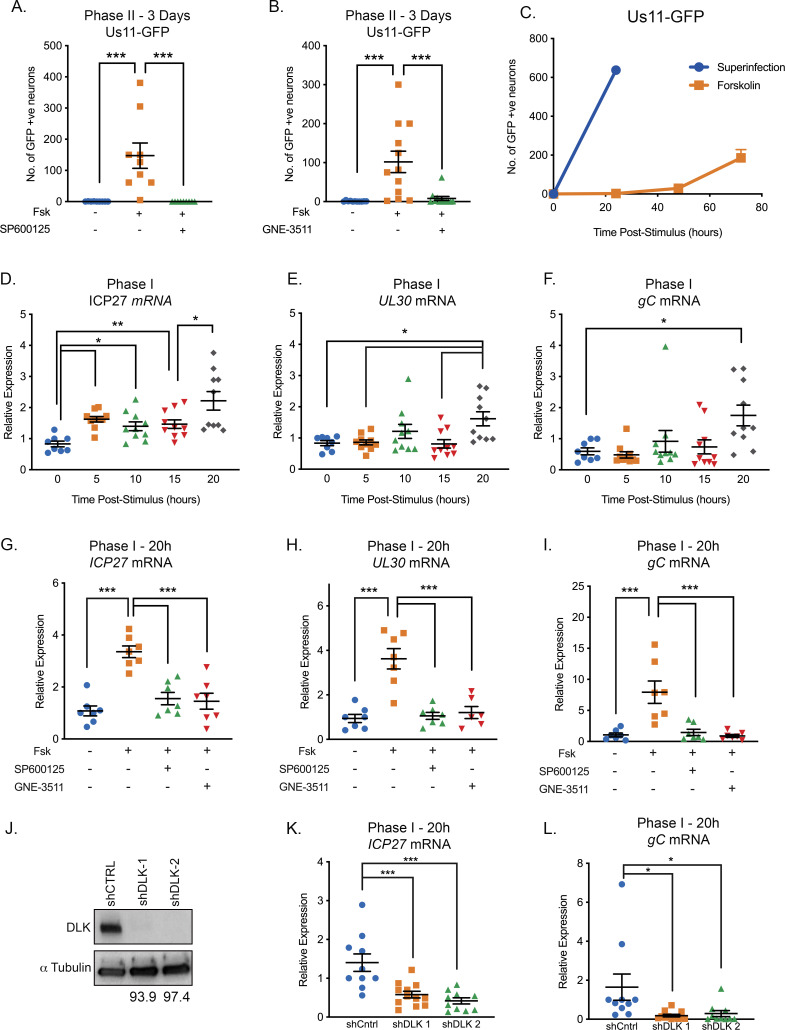
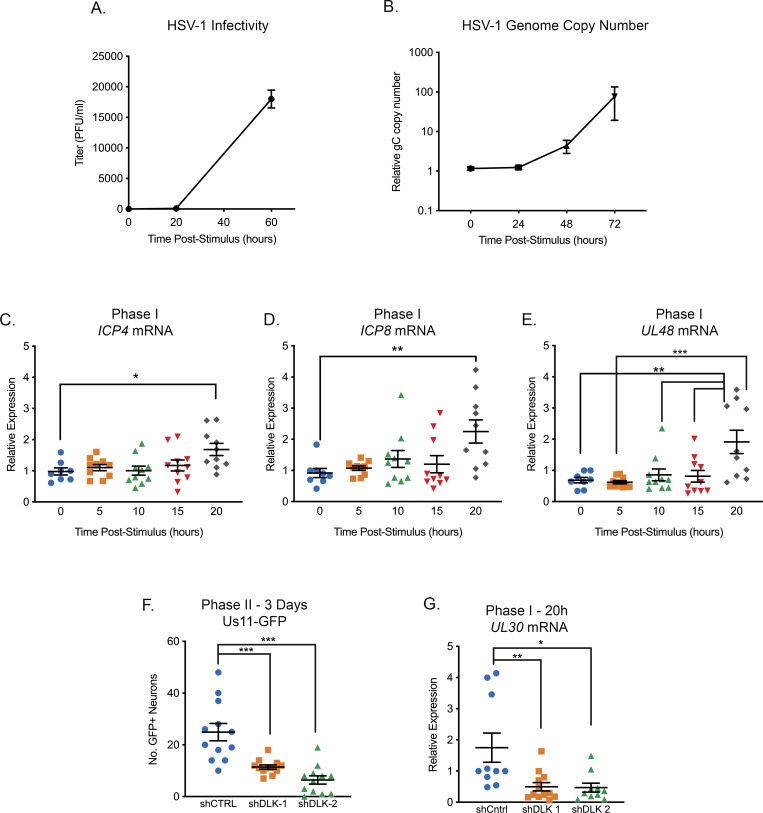
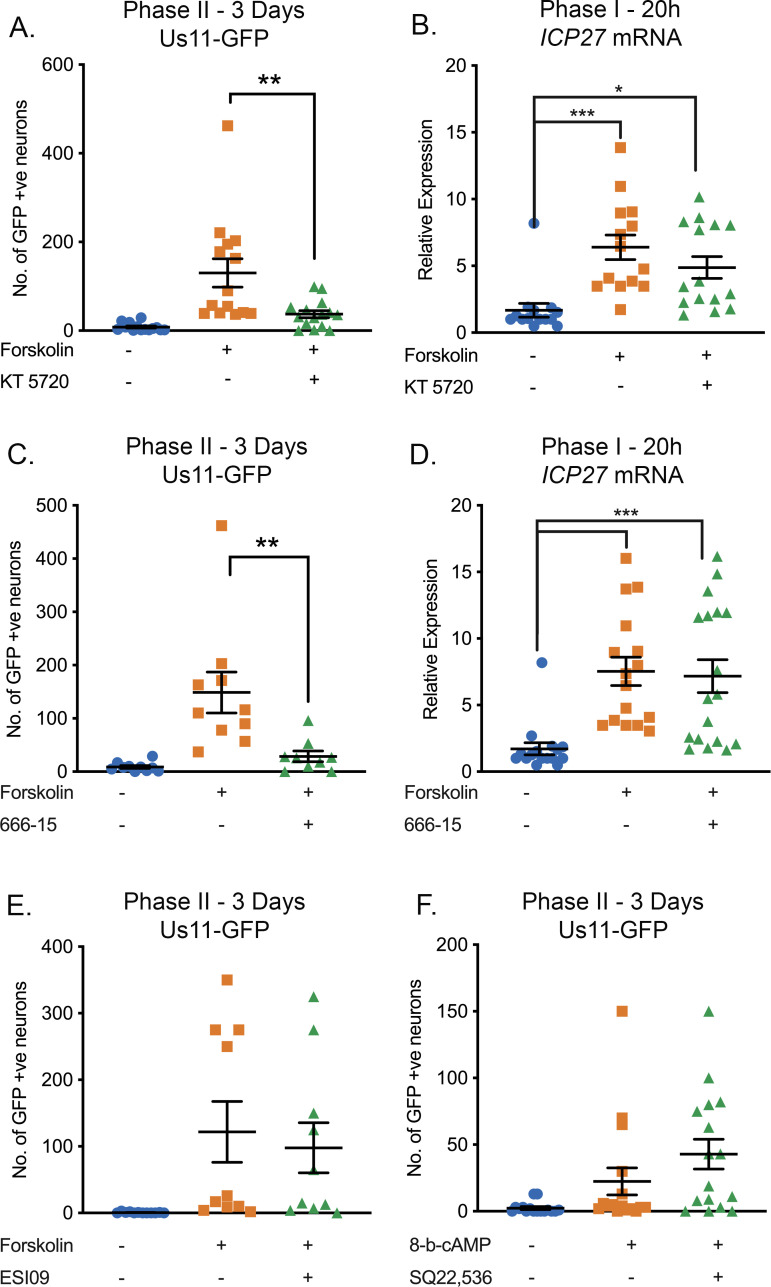
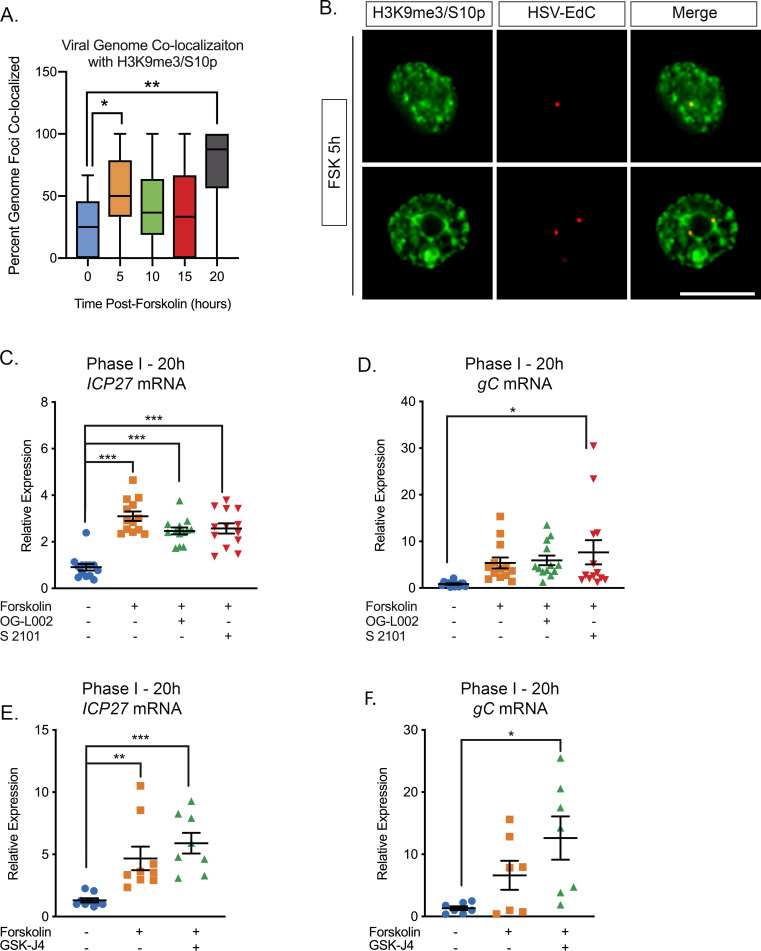
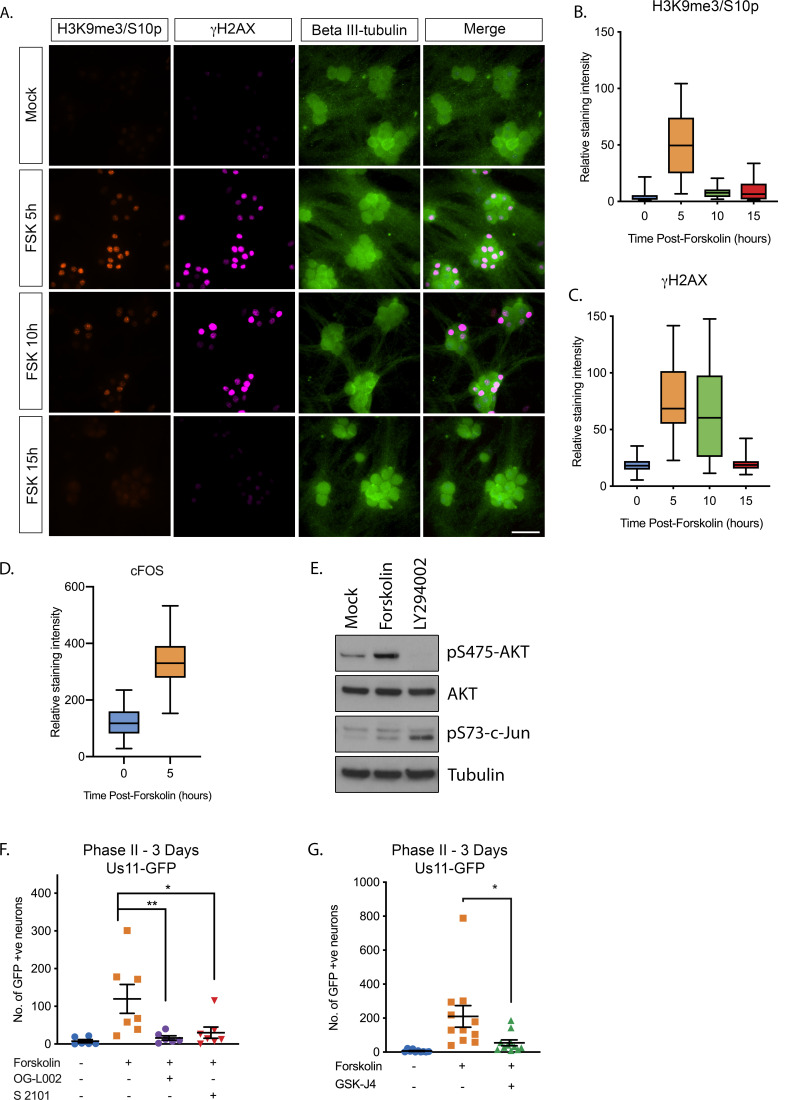
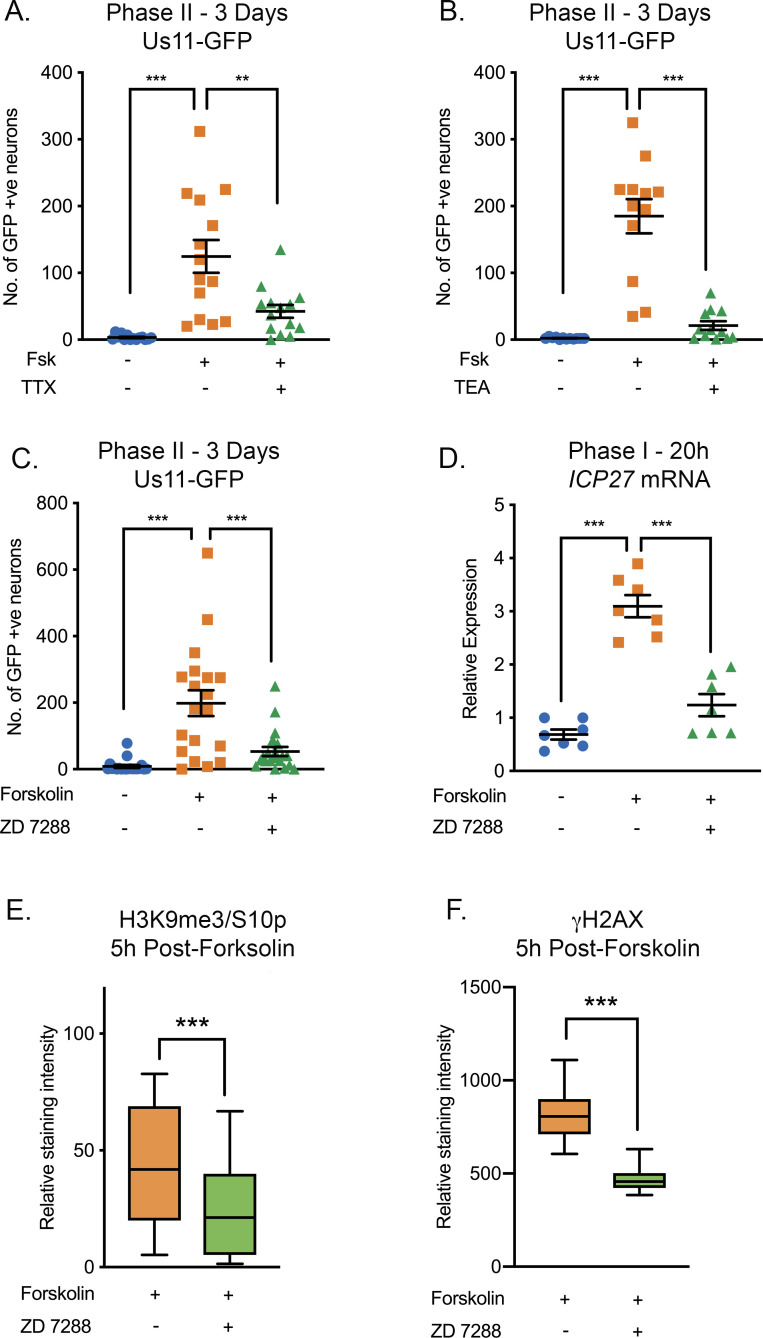
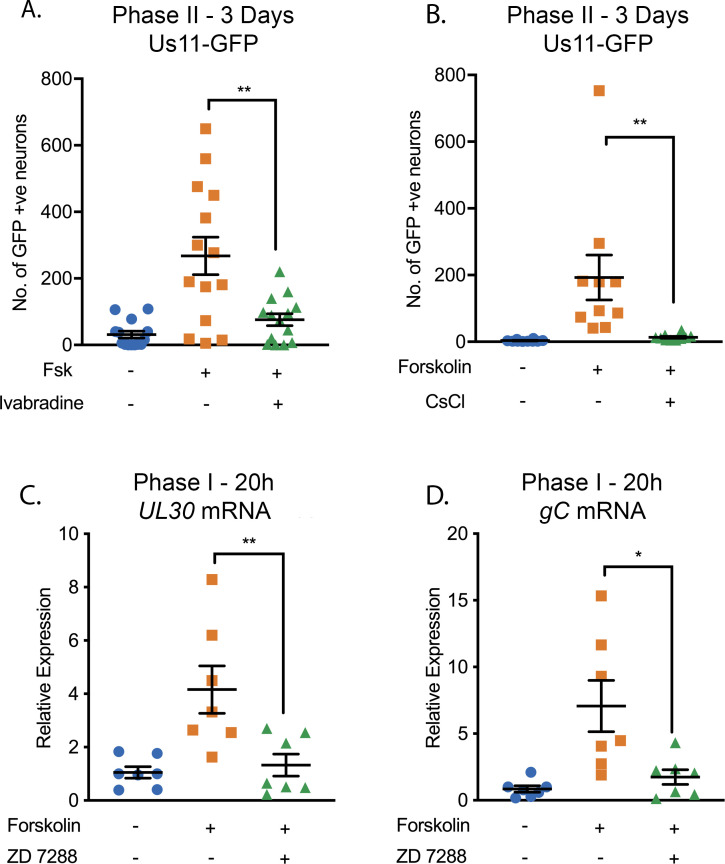
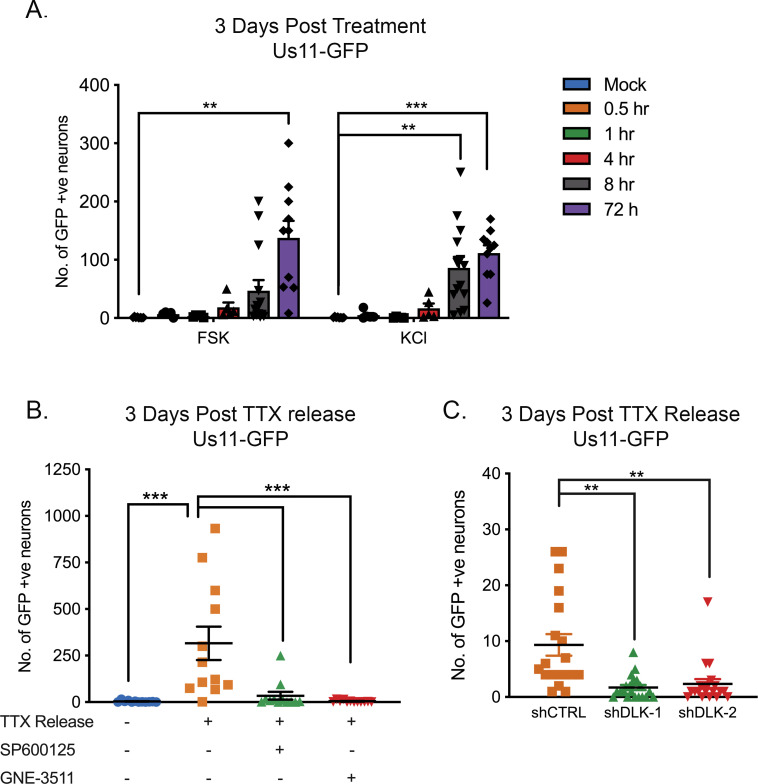
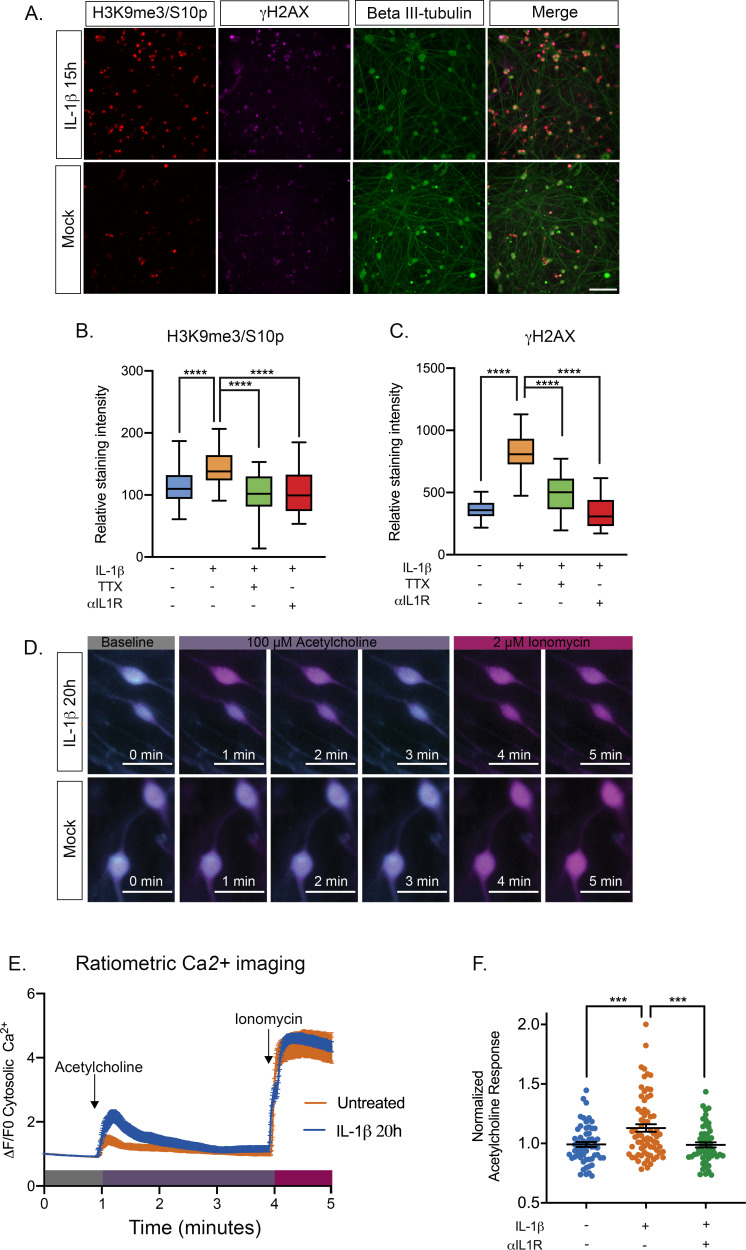
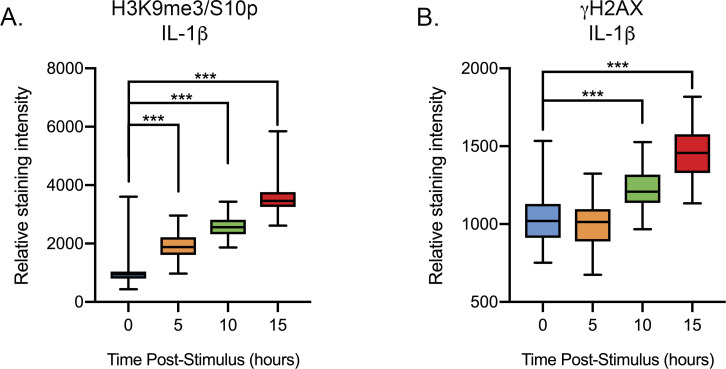
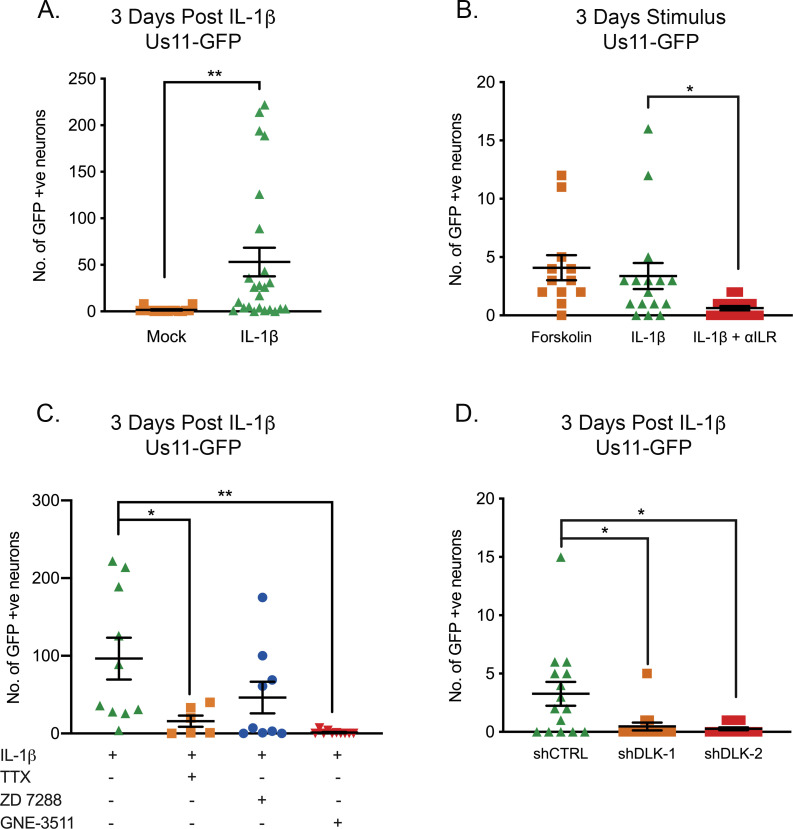
Herpes simplex virus-1 (HSV-1) establishes a latent infection in neurons and periodically reactivates to cause disease. The stimuli that trigger HSV-1 reactivation have not been fully elucidated. We demonstrate HSV-1 reactivation from latently infected mouse neurons induced by forskolin requires neuronal excitation. Stimuli that directly induce neurons to become hyperexcitable also induced HSV-1 reactivation. Forskolin-induced reactivation was dependent on the neuronal pathway of DLK/JNK activation and included an initial wave of viral gene expression that was independent of histone demethylase activity and linked to histone phosphorylation. IL-1β is released under conditions of stress, fever and UV exposure of the epidermis; all known triggers of clinical HSV reactivation. We found that IL-1β induced histone phosphorylation and increased the excitation in sympathetic neurons. Importantly, IL-1β triggered HSV-1 reactivation, which was dependent on DLK and neuronal excitability. Thus, HSV-1 co-opts an innate immune pathway resulting from IL-1 stimulation of neurons to induce reactivation.
Keywords: IL-1; dual leucine zipper kinase; epigenetics; herpes simplex virus; hyperexcitability; infectious disease; microbiology; mouse; neuroscience.
© 2020, Cuddy et al.
Conflict of interest statement
SC, AS, SD, PS, JS, PK, TD, MF, BD, CB, AC No competing interests declared
Figures
Similar articles
-
Ex Vivo Herpes Simplex Virus Reactivation Involves a Dual Leucine Zipper Kinase-Dependent Wave of Lytic Gene Expression That Is Independent of Histone Demethylase Activity and Viral Genome Synthesis.J Virol. 2022 Jun 22;96(12):e0047522. doi: 10.1128/jvi.00475-22. Epub 2022 May 23. J Virol. 2022. PMID: 35604215 Free PMC article.
-
DLK-Dependent Biphasic Reactivation of Herpes Simplex Virus Latency Established in the Absence of Antivirals.J Virol. 2022 Jun 22;96(12):e0050822. doi: 10.1128/jvi.00508-22. Epub 2022 May 24. J Virol. 2022. PMID: 35608347 Free PMC article.
-
Herpes Simplex Virus 1 Strains 17syn+ and KOS(M) Differ Greatly in Their Ability To Reactivate from Human Neurons In Vitro.J Virol. 2020 Jul 16;94(15):e00796-20. doi: 10.1128/JVI.00796-20. Print 2020 Jul 16. J Virol. 2020. PMID: 32461310 Free PMC article.
-
A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation.J Gen Virol. 2015 Jul;96(Pt 7):1581-602. doi: 10.1099/vir.0.000128. Epub 2015 Mar 20. J Gen Virol. 2015. PMID: 25794504 Free PMC article. Review.
-
[Mechanisms of herpes simplex virus latency and reactivation].Zhejiang Da Xue Xue Bao Yi Xue Ban. 2019 May 25;48(1):89-101. doi: 10.3785/j.issn.1008-9292.2019.02.14. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2019. PMID: 31102363 Free PMC article. Review. Chinese.
Cited by
-
Single-genome analysis reveals heterogeneous association of the Herpes Simplex Virus genome with H3K27me2 and the reader PHF20L1 following infection of human fibroblasts.bioRxiv [Preprint]. 2023 Dec 3:2023.12.03.569766. doi: 10.1101/2023.12.03.569766. bioRxiv. 2023. Update in: mBio. 2024 Apr 10;15(4):e0327823. doi: 10.1128/mbio.03278-23 PMID: 38076966 Free PMC article. Updated. Preprint.
-
c-Jun Signaling During Initial HSV-1 Infection Modulates Latency to Enhance Later Reactivation in addition to Directly Promoting the Progression to Full Reactivation.bioRxiv [Preprint]. 2023 Nov 10:2023.11.10.566462. doi: 10.1101/2023.11.10.566462. bioRxiv. 2023. Update in: J Virol. 2024 Feb 20;98(2):e0176423. doi: 10.1128/jvi.01764-23 PMID: 37986840 Free PMC article. Updated. Preprint.
-
A fur plucking model to study herpes simplex virus reactivation and recurrent disease.mSphere. 2024 Oct 29;9(10):e0078323. doi: 10.1128/msphere.00783-23. Epub 2024 Oct 9. mSphere. 2024. PMID: 39382285 Free PMC article.
-
Local Immune Control of Latent Herpes Simplex Virus Type 1 in Ganglia of Mice and Man.Front Immunol. 2021 Sep 15;12:723809. doi: 10.3389/fimmu.2021.723809. eCollection 2021. Front Immunol. 2021. PMID: 34603296 Free PMC article. Review.
-
Guideline for the Management Herpes Simplex 1 and Cosmetic Interventions.J Clin Aesthet Dermatol. 2021 Jun;14(6 Suppl 1):S11-S14. Epub 2021 Jun 1. J Clin Aesthet Dermatol. 2021. PMID: 34976293 Free PMC article.
References
-
- Alandijany T, Roberts APE, Conn KL, Loney C, McFarlane S, Orr A, Boutell C. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLOS Pathogens. 2018;14:e1006769. doi: 10.1371/journal.ppat.1006769. - DOI - PMC - PubMed
-
- Benboudjema L, Mulvey M, Gao Y, Pimplikar SW, Mohr I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. Journal of Virology. 2003;77:9192–9203. doi: 10.1128/JVI.77.17.9192-9203.2003. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
