

Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases
- PMID: 33240899
- PMCID: PMC7680862
- DOI: 10.3389/fcell.2020.604240
Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases
Abstract
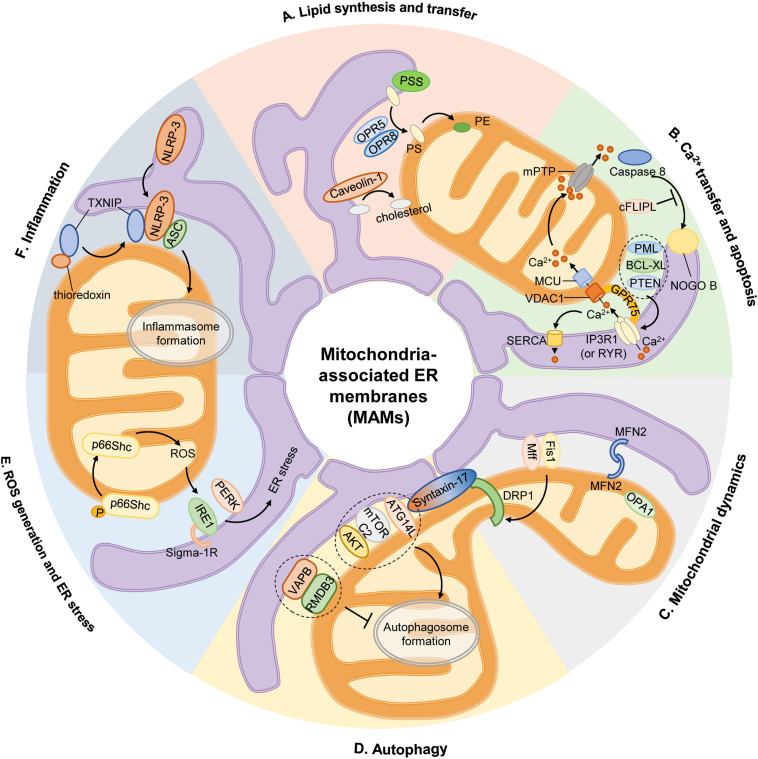
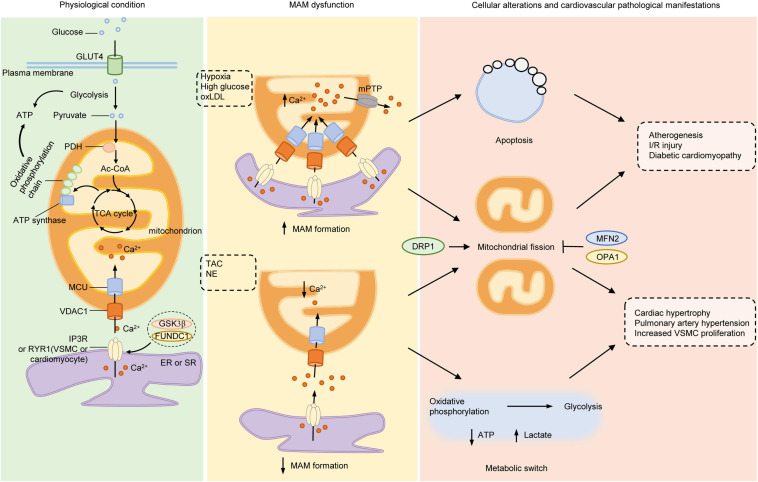
The endoplasmic reticulum (ER) and mitochondria are physically connected to form dedicated structural domains known as mitochondria-associated ER membranes (MAMs), which participate in fundamental biological processes, including lipid and calcium (Ca2+) homeostasis, mitochondrial dynamics and other related cellular behaviors such as autophagy, ER stress, inflammation and apoptosis. Many studies have proved the importance of MAMs in maintaining the normal function of both organelles, and the abnormal amount, structure or function of MAMs is related to the occurrence of cardiovascular diseases. Here, we review the knowledge regarding the components of MAMs according to their different functions and the specific roles of MAMs in cardiovascular physiology and pathophysiology, focusing on some highly prevalent cardiovascular diseases, including ischemia-reperfusion, diabetic cardiomyopathy, heart failure, pulmonary arterial hypertension and systemic vascular diseases. Finally, we summarize the possible mechanisms of MAM in cardiovascular diseases and put forward some obstacles in the understanding of MAM function we may encounter.
Keywords: SR-mitochondrial contact; cardiovascular diseases; metabolic transition; mitochondria-associated ER membrane; mitochondrial bioenergetics.
Copyright © 2020 Gao, Yan and Zhu.
Figures
Similar articles
-
Cal'MAM'ity at the Endoplasmic Reticulum-Mitochondrial Interface: A Potential Therapeutic Target for Neurodegeneration and Human Immunodeficiency Virus-Associated Neurocognitive Disorders.Front Neurosci. 2021 Oct 21;15:715945. doi: 10.3389/fnins.2021.715945. eCollection 2021. Front Neurosci. 2021. PMID: 34744606 Free PMC article. Review.
-
Potential Roles of Mitochondria-Associated ER Membranes (MAMs) in Traumatic Brain Injury.Cell Mol Neurobiol. 2017 Nov;37(8):1349-1357. doi: 10.1007/s10571-017-0484-2. Epub 2017 Mar 21. Cell Mol Neurobiol. 2017. PMID: 28324201 Review.
-
The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance.Front Endocrinol (Lausanne). 2020 Nov 23;11:592129. doi: 10.3389/fendo.2020.592129. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 33329397 Free PMC article. Review.
-
ER-to-mitochondria miscommunication and metabolic diseases.Biochim Biophys Acta. 2015 Oct;1852(10 Pt A):2096-105. doi: 10.1016/j.bbadis.2015.07.011. Epub 2015 Jul 11. Biochim Biophys Acta. 2015. PMID: 26171812 Review.
-
Dialogue between mitochondria and endoplasmic reticulum-potential therapeutic targets for age-related cardiovascular diseases.Front Pharmacol. 2024 Jun 13;15:1389202. doi: 10.3389/fphar.2024.1389202. eCollection 2024. Front Pharmacol. 2024. PMID: 38939842 Free PMC article. Review.
Cited by
-
The Downregulation of ADAM17 Exerts Protective Effects against Cardiac Fibrosis by Regulating Endoplasmic Reticulum Stress and Mitophagy.Oxid Med Cell Longev. 2021 May 6;2021:5572088. doi: 10.1155/2021/5572088. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34035876 Free PMC article.
-
Diabetic cardiomyopathy: a brief summary on lipid toxicity.ESC Heart Fail. 2023 Apr;10(2):776-790. doi: 10.1002/ehf2.14224. Epub 2022 Nov 11. ESC Heart Fail. 2023. PMID: 36369594 Free PMC article. Review.
-
Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest.Antioxidants (Basel). 2021 Mar 8;10(3):406. doi: 10.3390/antiox10030406. Antioxidants (Basel). 2021. PMID: 33800427 Free PMC article. Review.
-
Mitochondria-Endoplasmic Reticulum Contacts: The Promising Regulators in Diabetic Cardiomyopathy.Oxid Med Cell Longev. 2022 Apr 11;2022:2531458. doi: 10.1155/2022/2531458. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 35450404 Free PMC article. Review.
-
Mitochondria-endoplasmic reticulum contacts in sepsis-induced myocardial dysfunction.Front Cell Dev Biol. 2022 Nov 24;10:1036225. doi: 10.3389/fcell.2022.1036225. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36506093 Free PMC article. Review.
References
-
- Akhmedov A., Montecucco F., Braunersreuther V., Camici G. G., Jakob P., Reiner M. F., et al. (2015). Genetic deletion of the adaptor protein p66Shc increases susceptibility to short-term ischaemic myocardial injury via intracellular salvage pathways. Eur. Heart J. 36 516a-526a. 10.1093/eurheartj/ehu400 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous