

Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer's disease
- PMID: 33075193
- PMCID: PMC7983919
- DOI: 10.1002/alz.12192
Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer's disease
Abstract
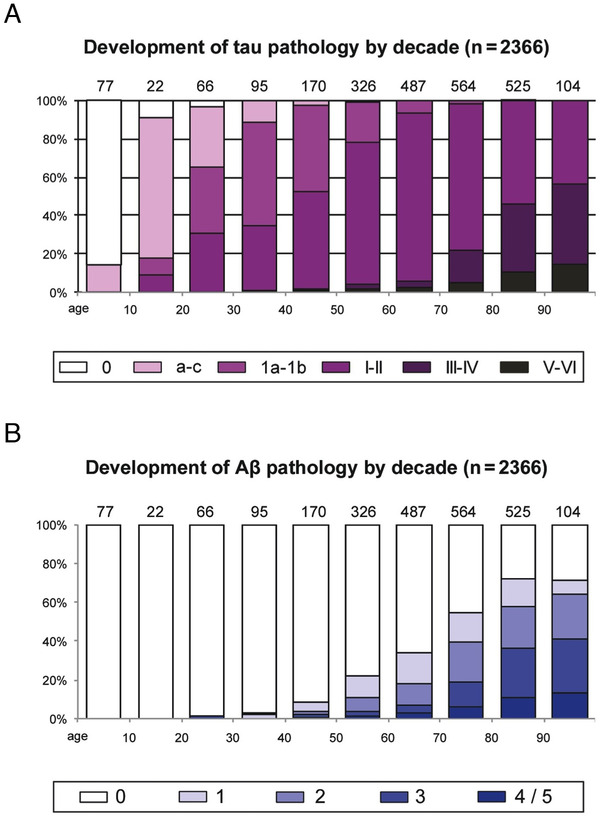
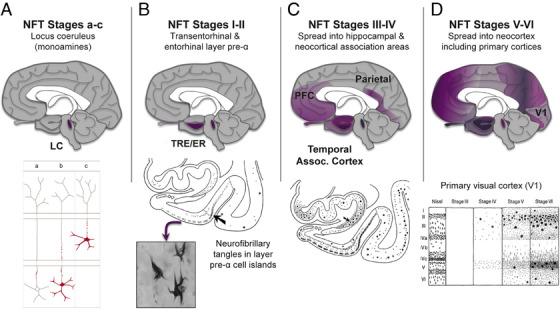
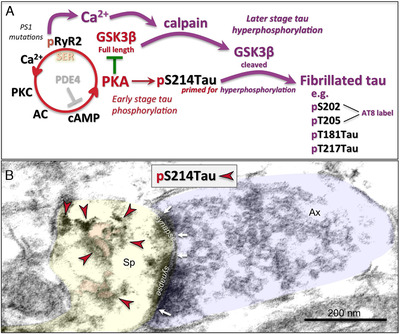
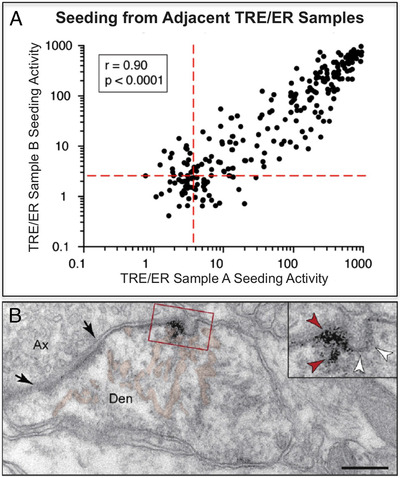
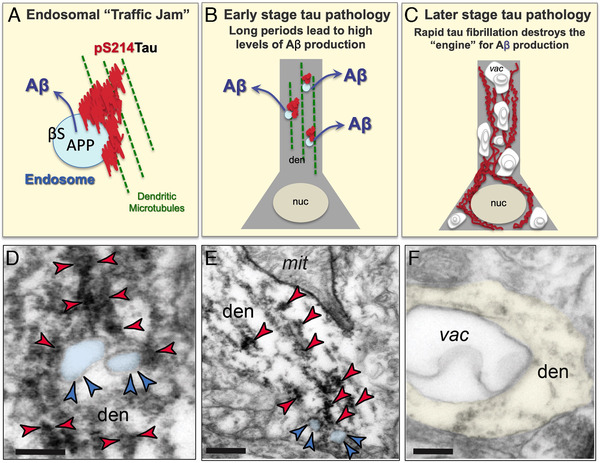
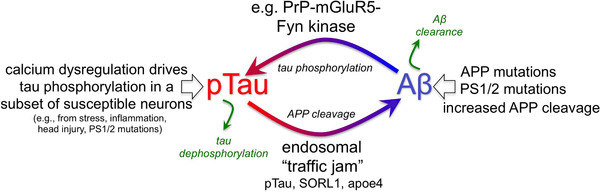
The etiology of the common, sporadic form of Alzheimer's disease (sAD) is unknown. We hypothesize that tau pathology within select projection neurons with susceptible microenvironments can initiate sAD. This postulate rests on extensive data demonstrating that in human brains tau pathology appears about a decade before the formation of Aβ plaques (Aβps), especially targeting glutamate projection neurons in the association cortex. Data from aging rhesus monkeys show abnormal tau phosphorylation within vulnerable neurons, associated with calcium dysregulation. Abnormally phosphorylated tau (pTau) on microtubules traps APP-containing endosomes, which can increase Aβ production. As Aβ oligomers increase abnormal phosphorylation of tau, this would drive vicious cycles leading to sAD pathology over a long lifespan, with genetic and environmental factors that may accelerate pathological events. This hypothesis could be testable in the aged monkey association cortex that naturally expresses characteristics capable of promoting and sustaining abnormal tau phosphorylation and Aβ production.
Keywords: abnormally phosphorylated tau; association cortex; calcium; rhesus monkey; sporadic Alzheimer's disease; tau seeding; β-amyloid.
© 2020 The Authors. Alzheimer's & Dementia published by Wiley Periodicals LLC on behalf of Alzheimer's Association.
Conflict of interest statement
Amy F.T. Arnsten and Yale receive royalties from the USA sales of Intuniv (extended release guanfacine). They do not receive royalties from non‐USA or generic sales of Intuniv. The other authors have no actual or potential conflicts of interest to declare.
Figures
Similar articles
-
Studies of aging nonhuman primates illuminate the etiology of early-stage Alzheimer's-like neuropathology: An evolutionary perspective.Am J Primatol. 2021 Nov;83(11):e23254. doi: 10.1002/ajp.23254. Epub 2021 May 7. Am J Primatol. 2021. PMID: 33960505 Free PMC article. Review.
-
Brains of rhesus monkeys display Aβ deposits and glial pathology while lacking Aβ dimers and other Alzheimer's pathologies.Aging Cell. 2019 Aug;18(4):e12978. doi: 10.1111/acel.12978. Epub 2019 Jun 4. Aging Cell. 2019. PMID: 31165579 Free PMC article.
-
Neurons derived from sporadic Alzheimer's disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation.Alzheimers Res Ther. 2017 Dec 1;9(1):90. doi: 10.1186/s13195-017-0317-z. Alzheimers Res Ther. 2017. PMID: 29191219 Free PMC article.
-
Amyloid-beta and phosphorylated tau in post-mortem Alzheimer's disease retinas.Acta Neuropathol Commun. 2018 Dec 28;6(1):147. doi: 10.1186/s40478-018-0650-x. Acta Neuropathol Commun. 2018. PMID: 30593285 Free PMC article.
-
Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies.Curr Alzheimer Res. 2005 Jan;2(1):3-18. doi: 10.2174/1567205052772713. Curr Alzheimer Res. 2005. PMID: 15977985 Review.
Cited by
-
Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer's Disease and Identifying Promising Drug Targets.Biomolecules. 2022 Nov 11;12(11):1676. doi: 10.3390/biom12111676. Biomolecules. 2022. PMID: 36421690 Free PMC article. Review.
-
The Vascular-Immune Hypothesis of Alzheimer's Disease.Biomedicines. 2023 Jan 30;11(2):408. doi: 10.3390/biomedicines11020408. Biomedicines. 2023. PMID: 36830944 Free PMC article. Review.
-
What's the cut-point?: a systematic investigation of tau PET thresholding methods.Alzheimers Res Ther. 2022 Apr 5;14(1):49. doi: 10.1186/s13195-022-00986-w. Alzheimers Res Ther. 2022. PMID: 35382866 Free PMC article. Review.
-
Induction of Brain Insulin Resistance and Alzheimer's Molecular Changes by Western Diet.Int J Mol Sci. 2022 Apr 25;23(9):4744. doi: 10.3390/ijms23094744. Int J Mol Sci. 2022. PMID: 35563135 Free PMC article.
-
Localization of PDE4D, HCN1 channels, and mGluR3 in rhesus macaque entorhinal cortex may confer vulnerability in Alzheimer's disease.Cereb Cortex. 2023 Dec 9;33(24):11501-11516. doi: 10.1093/cercor/bhad382. Cereb Cortex. 2023. PMID: 37874022 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
