

SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling
- PMID: 33045063
- PMCID: PMC7549316
- DOI: 10.1084/jem.20201414
SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling
Abstract
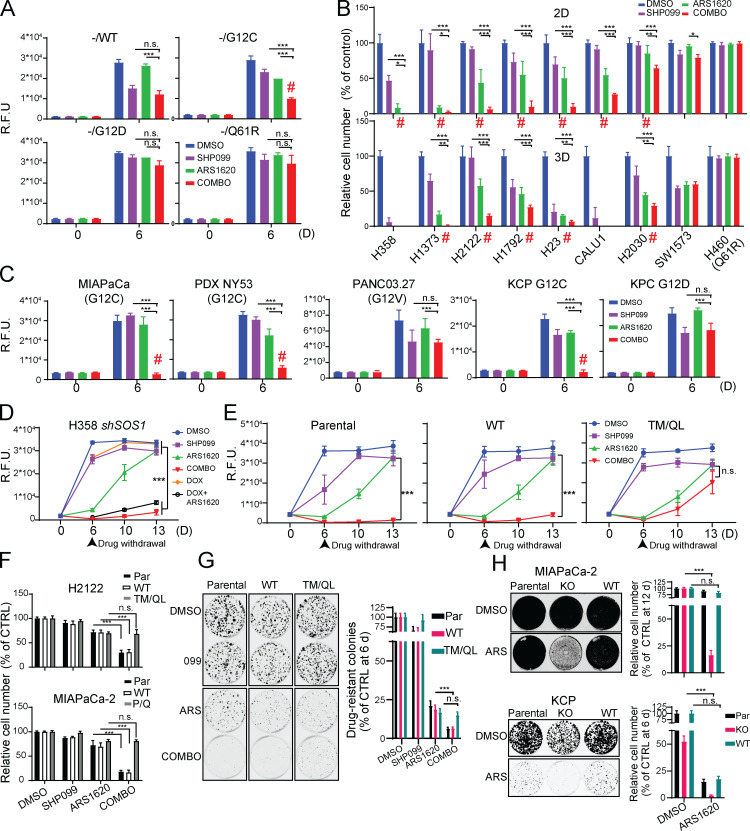
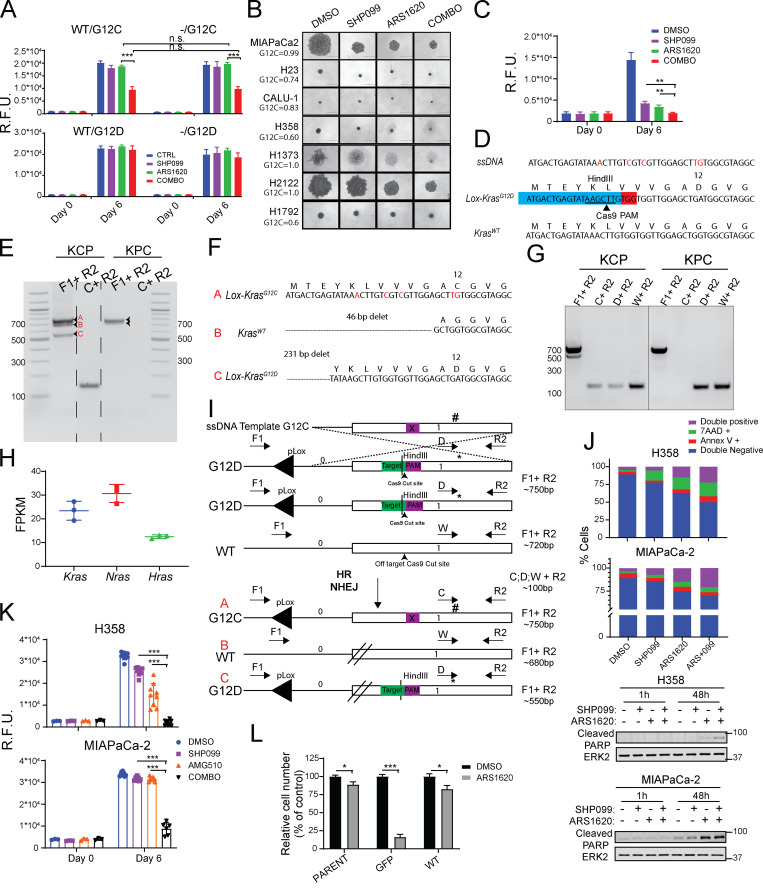
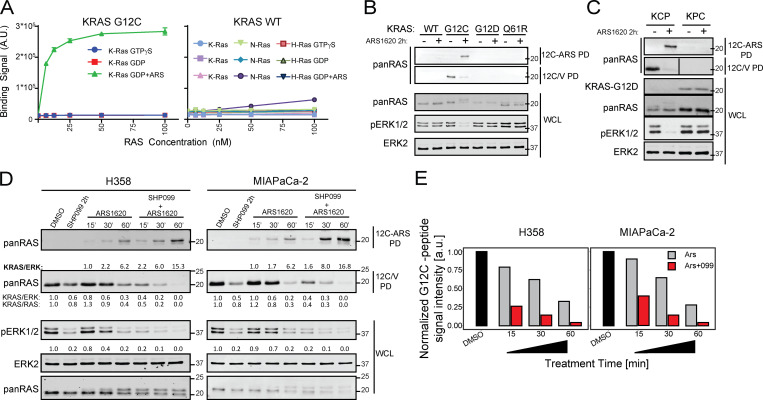
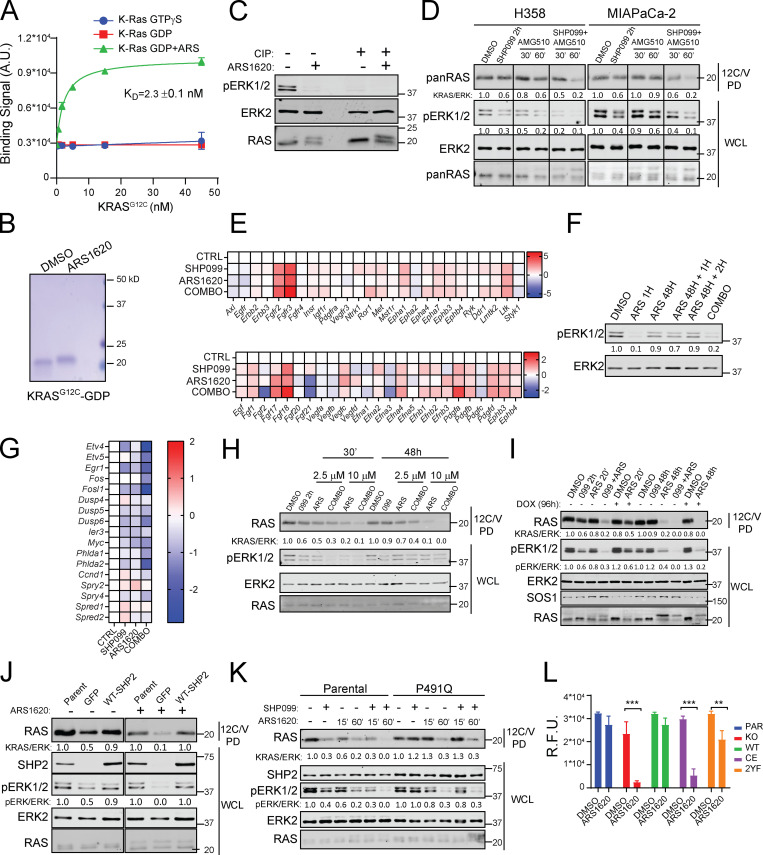
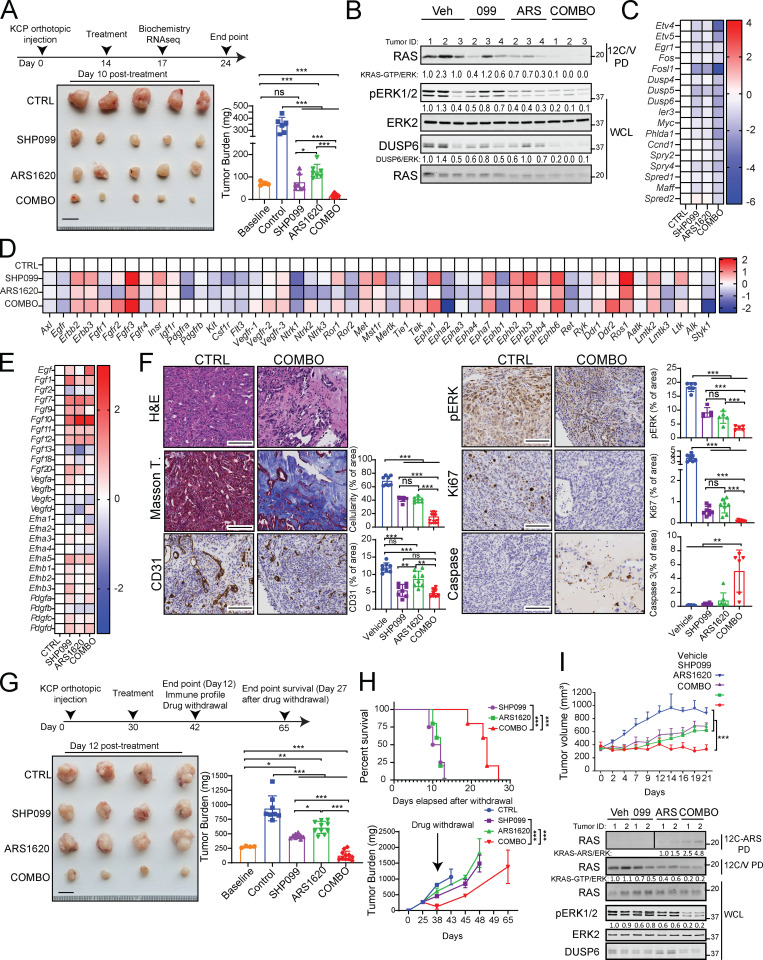
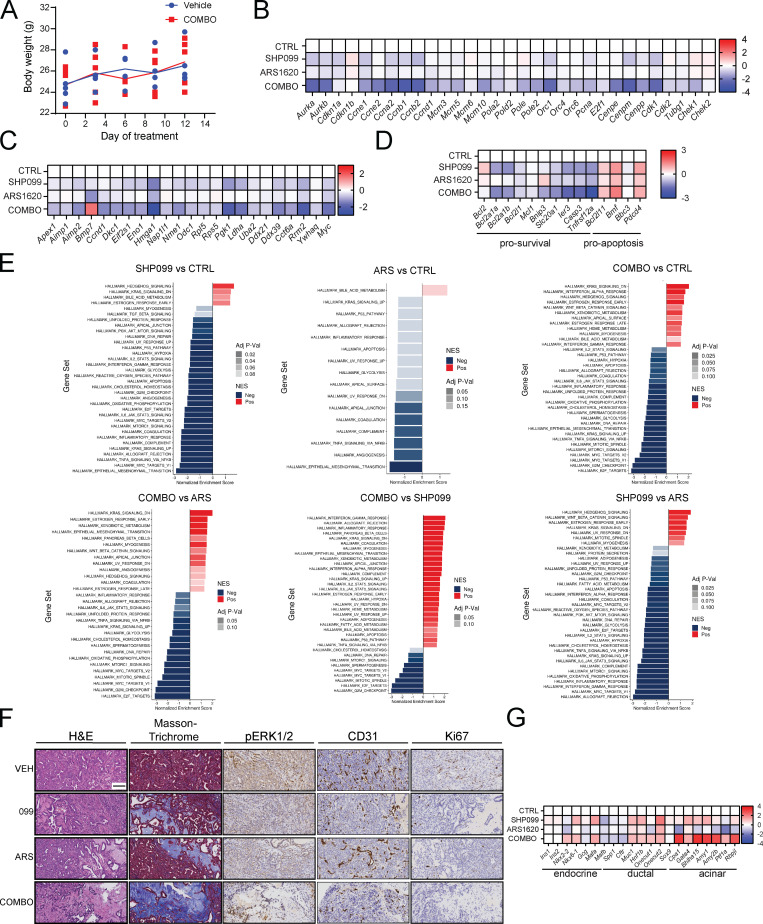
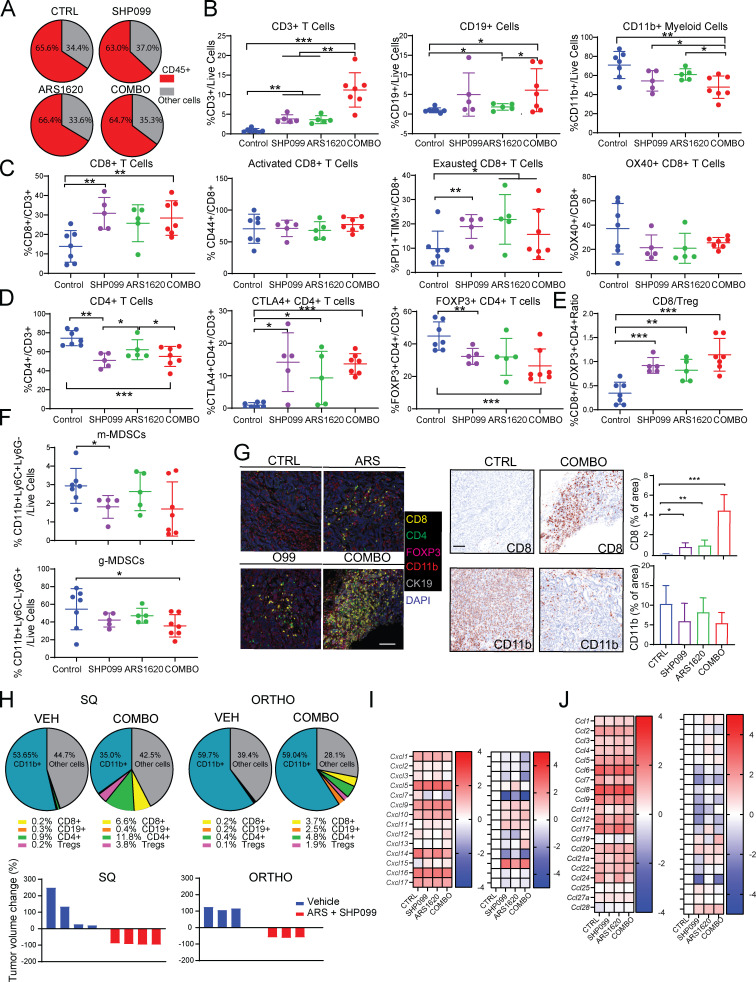
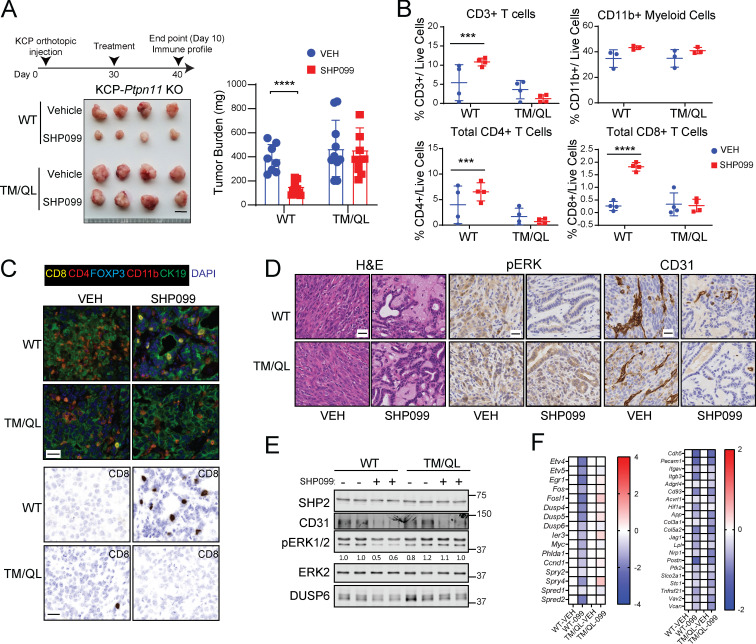
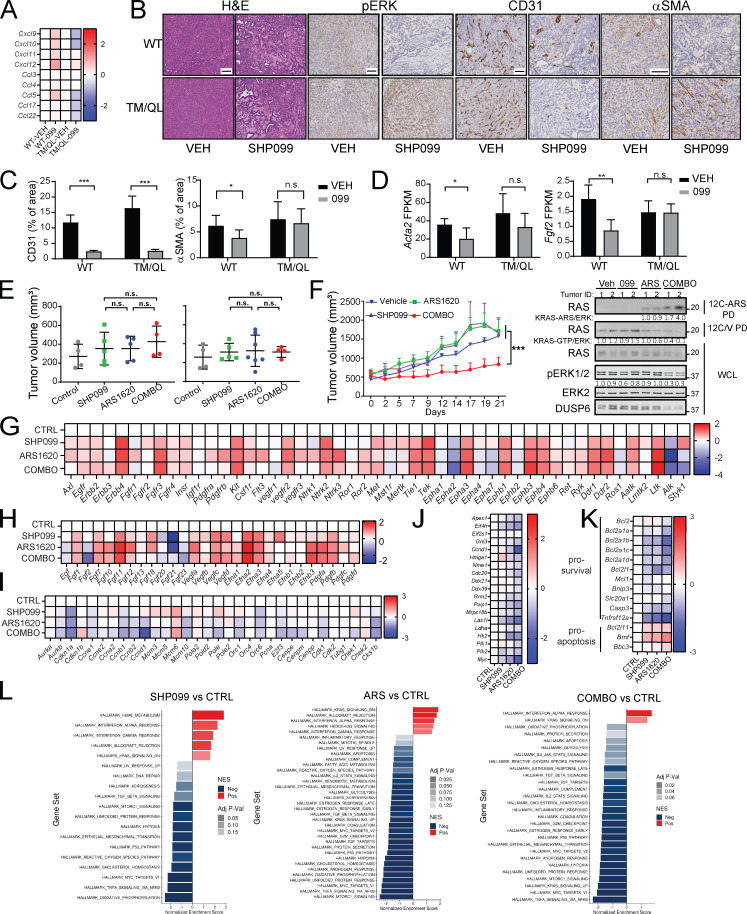
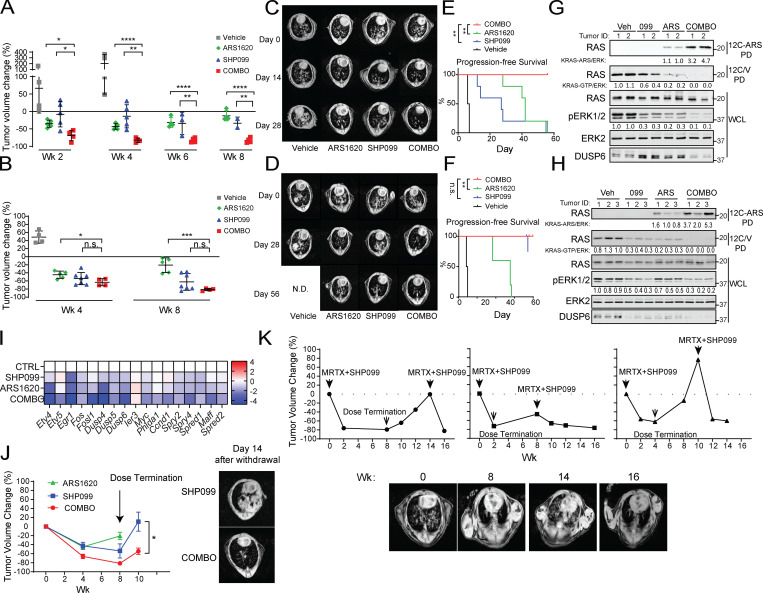
KRAS is the most frequently mutated human oncogene, and KRAS inhibition has been a longtime goal. Recently, inhibitors were developed that bind KRASG12C-GDP and react with Cys-12 (G12C-Is). Using new affinity reagents to monitor KRASG12C activation and inhibitor engagement, we found that an SHP2 inhibitor (SHP2-I) increases KRAS-GDP occupancy, enhancing G12C-I efficacy. The SHP2-I abrogated RTK feedback signaling and adaptive resistance to G12C-Is in vitro, in xenografts, and in syngeneic KRASG12C-mutant pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC). SHP2-I/G12C-I combination evoked favorable but tumor site-specific changes in the immune microenvironment, decreasing myeloid suppressor cells, increasing CD8+ T cells, and sensitizing tumors to PD-1 blockade. Experiments using cells expressing inhibitor-resistant SHP2 showed that SHP2 inhibition in PDAC cells is required for PDAC regression and remodeling of the immune microenvironment but revealed direct inhibitory effects on tumor angiogenesis and vascularity. Our results demonstrate that SHP2-I/G12C-I combinations confer a substantial survival benefit in PDAC and NSCLC and identify additional potential combination strategies.
© 2020 Fedele et al.
Conflict of interest statement
Disclosures: C.J.R. Foster reported grants from the National Institutes of Health during the conduct of the study. K-K. Wong reported "other" from G1 Therapeutics, Zentalis Therapeutics, and Epiphanes Therapeutics outside the submitted work, and has consulting/sponsored research agreements with the following: AstraZeneca, Janssen, Pfizer, Novartis, Merck, Ono, and Array (consulting and sponsored research); MedImmune, Mirati (which developed MRTX 1257), Takeda, TargImmune, and BMS (sponsored research only). B.G. Neel reported "other" from Navire Pharma, Northern Biologics, Ltd, Arvinas, Inc, Regeneron, Amgen, Inc, Mirati Therapeutics, Gilead Therapeutics, and Moderna outside the submitted work. In addition, B.G. Neel has a patent to PCT 63031457 pending. No other disclosures were reported.
Figures




Similar articles
-
CRISPR Screening Identifies Mechanisms of Resistance to KRASG12C and SHP2 Inhibitor Combinations in Non-Small Cell Lung Cancer.Cancer Res. 2023 Dec 15;83(24):4130-4141. doi: 10.1158/0008-5472.CAN-23-1127. Cancer Res. 2023. PMID: 37934115 Free PMC article.
-
High SHP2 expression determines the efficacy of PD-1/PD-L1 inhibitors in advanced KRAS mutant non-small cell lung cancer.Thorac Cancer. 2021 Oct;12(19):2564-2573. doi: 10.1111/1759-7714.14137. Epub 2021 Sep 6. Thorac Cancer. 2021. PMID: 34490728 Free PMC article.
-
Combining RAS(ON) G12C-selective inhibitor with SHP2 inhibition sensitises lung tumours to immune checkpoint blockade.Nat Commun. 2024 Sep 25;15(1):8146. doi: 10.1038/s41467-024-52324-3. Nat Commun. 2024. PMID: 39322643 Free PMC article.
-
Targeting KRASp.G12C Mutation in Advanced Non-Small Cell Lung Cancer: a New Era Has Begun.Curr Treat Options Oncol. 2022 Dec;23(12):1699-1720. doi: 10.1007/s11864-022-01033-4. Epub 2022 Nov 17. Curr Treat Options Oncol. 2022. PMID: 36394791 Review.
-
Unveiling the role of KRAS in tumor immune microenvironment.Biomed Pharmacother. 2024 Feb;171:116058. doi: 10.1016/j.biopha.2023.116058. Epub 2024 Jan 2. Biomed Pharmacother. 2024. PMID: 38171240 Review.
Cited by
-
Co-targeting SOS1 enhances the antitumor effects of KRASG12C inhibitors by addressing intrinsic and acquired resistance.Nat Cancer. 2024 Sep;5(9):1352-1370. doi: 10.1038/s43018-024-00800-6. Epub 2024 Aug 5. Nat Cancer. 2024. PMID: 39103541 Free PMC article.
-
Mechanisms of resistance to KRASG12C inhibitors in KRASG12C-mutated non-small cell lung cancer.Front Oncol. 2024 Sep 5;14:1328728. doi: 10.3389/fonc.2024.1328728. eCollection 2024. Front Oncol. 2024. PMID: 39301544 Free PMC article. Review.
-
Targeted Therapies for Kirsten Rat Sarcoma (KRAS) G12C Mutant Metastatic Non-Small-Cell Lung Cancers.Cancers (Basel). 2023 Nov 25;15(23):5582. doi: 10.3390/cancers15235582. Cancers (Basel). 2023. PMID: 38067288 Free PMC article. Review.
-
WWP1 inhibition increases SHP2 inhibitor efficacy in colorectal cancer.NPJ Precis Oncol. 2024 Jul 16;8(1):144. doi: 10.1038/s41698-024-00650-6. NPJ Precis Oncol. 2024. PMID: 39014007 Free PMC article.
-
Genomic Landscapes and Hallmarks of Mutant RAS in Human Cancers.Cancer Res. 2022 Nov 2;82(21):4058-4078. doi: 10.1158/0008-5472.CAN-22-1731. Cancer Res. 2022. PMID: 36074020 Free PMC article.
References
-
- Anderson G.R., Winter P.S., Lin K.H., Nussbaum D.P., Cakir M., Stein E.M., Soderquist R.S., Crawford L., Leeds J.C., Newcomb R., et al. . 2017. A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Rep. 20:999–1015. 10.1016/j.celrep.2017.07.006 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
