

Notch Signaling in Breast Cancer: A Role in Drug Resistance
- PMID: 33003540
- PMCID: PMC7601482
- DOI: 10.3390/cells9102204
Notch Signaling in Breast Cancer: A Role in Drug Resistance
Abstract
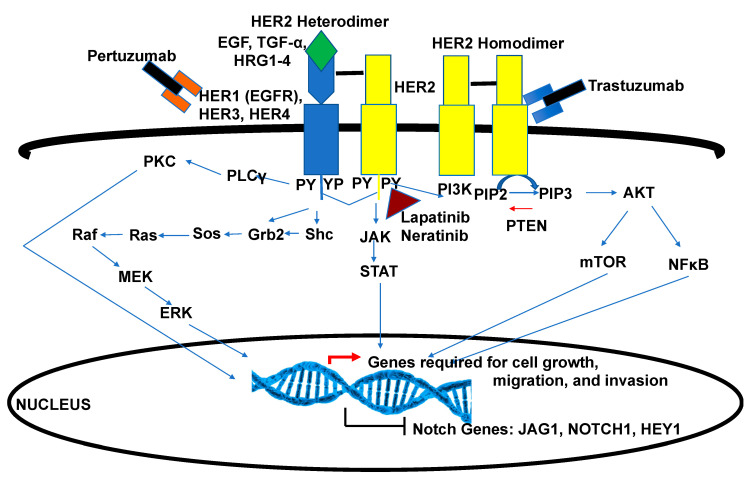
Breast cancer is a heterogeneous disease that can be subdivided into unique molecular subtypes based on protein expression of the Estrogen Receptor, Progesterone Receptor, and/or the Human Epidermal Growth Factor Receptor 2. Therapeutic approaches are designed to inhibit these overexpressed receptors either by endocrine therapy, targeted therapies, or combinations with cytotoxic chemotherapy. However, a significant percentage of breast cancers are inherently resistant or acquire resistance to therapies, and mechanisms that promote resistance remain poorly understood. Notch signaling is an evolutionarily conserved signaling pathway that regulates cell fate, including survival and self-renewal of stem cells, proliferation, or differentiation. Deregulation of Notch signaling promotes resistance to targeted or cytotoxic therapies by enriching of a small population of resistant cells, referred to as breast cancer stem cells, within the bulk tumor; enhancing stem-like features during the process of de-differentiation of tumor cells; or promoting epithelial to mesenchymal transition. Preclinical studies have shown that targeting the Notch pathway can prevent or reverse resistance through reduction or elimination of breast cancer stem cells. However, Notch inhibitors have yet to be clinically approved for the treatment of breast cancer, mainly due to dose-limiting gastrointestinal toxicity. In this review, we discuss potential mechanisms of Notch-mediated resistance in breast cancer cells and breast cancer stem cells, and various methods of targeting Notch through γ-secretase inhibitors, Notch signaling biologics, or transcriptional inhibitors. We also discuss future plans for identification of novel Notch-targeted therapies, in order to reduce toxicity and improve outcomes for women with resistant breast cancer.
Keywords: Notch; breast cancer; cancer stem cells; resistance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation.Anticancer Res. 2010 Oct;30(10):3853-67. Anticancer Res. 2010. PMID: 21036696
-
Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells.Cancer Lett. 2013 Nov 28;341(1):41-5. doi: 10.1016/j.canlet.2013.08.027. Epub 2013 Aug 21. Cancer Lett. 2013. PMID: 23973264 Review.
-
Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells.Breast Cancer Res. 2014 Jun 11;16(3):R62. doi: 10.1186/bcr3675. Breast Cancer Res. 2014. PMID: 24919951 Free PMC article.
-
Blocking the NOTCH pathway can inhibit the growth of CD133-positive A549 cells and sensitize to chemotherapy.Biochem Biophys Res Commun. 2014 Feb 21;444(4):670-5. doi: 10.1016/j.bbrc.2014.01.164. Epub 2014 Feb 3. Biochem Biophys Res Commun. 2014. PMID: 24502949
-
Rational targeting of Notch signaling in cancer.Oncogene. 2008 Sep 1;27(38):5124-31. doi: 10.1038/onc.2008.226. Oncogene. 2008. PMID: 18758481 Review.
Cited by
-
Cancer Stem Cells-The Insight into Non-Coding RNAs.Cells. 2022 Nov 21;11(22):3699. doi: 10.3390/cells11223699. Cells. 2022. PMID: 36429127 Free PMC article. Review.
-
Advances of nanotechnology applied to cancer stem cells.World J Stem Cells. 2023 Jun 26;15(6):514-529. doi: 10.4252/wjsc.v15.i6.514. World J Stem Cells. 2023. PMID: 37424953 Free PMC article. Review.
-
Prognostic and predictive impact of NOTCH1 in early breast cancer.Breast Cancer Res Treat. 2025 Jan;209(1):27-38. doi: 10.1007/s10549-024-07444-1. Epub 2024 Aug 17. Breast Cancer Res Treat. 2025. PMID: 39153127 Free PMC article.
-
Reciprocal inhibition of NOTCH and SOX2 shapes tumor cell plasticity and therapeutic escape in triple-negative breast cancer.EMBO Mol Med. 2024 Dec;16(12):3184-3217. doi: 10.1038/s44321-024-00161-8. Epub 2024 Oct 30. EMBO Mol Med. 2024. PMID: 39478150 Free PMC article.
-
Cancer Stem Cells and the Tumor Microenvironment: Targeting the Critical Crosstalk through Nanocarrier Systems.Stem Cell Rev Rep. 2022 Oct;18(7):2209-2233. doi: 10.1007/s12015-022-10426-9. Epub 2022 Jul 25. Stem Cell Rev Rep. 2022. PMID: 35876959 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
