

Innate immunity during SARS-CoV-2: evasion strategies and activation trigger hypoxia and vascular damage
- PMID: 32978971
- PMCID: PMC7537271
- DOI: 10.1111/cei.13523
Innate immunity during SARS-CoV-2: evasion strategies and activation trigger hypoxia and vascular damage
Abstract
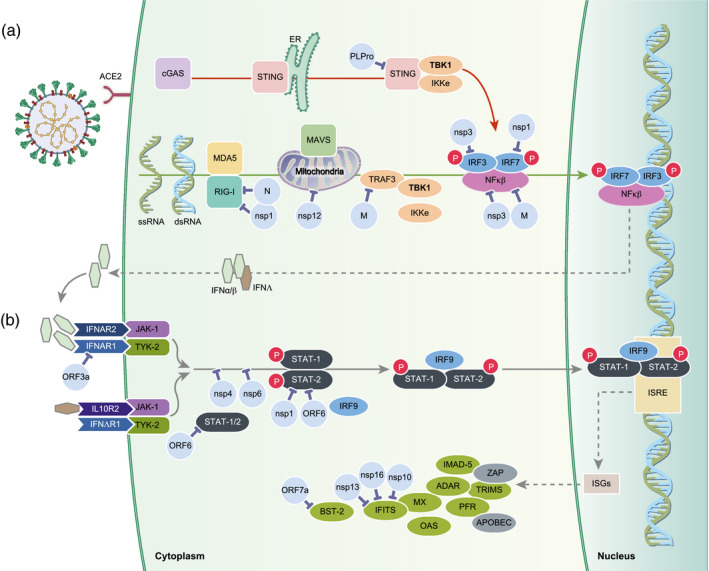
Innate immune sensing of viral molecular patterns is essential for development of antiviral responses. Like many viruses, SARS-CoV-2 has evolved strategies to circumvent innate immune detection, including low cytosine-phosphate-guanosine (CpG) levels in the genome, glycosylation to shield essential elements including the receptor-binding domain, RNA shielding and generation of viral proteins that actively impede anti-viral interferon responses. Together these strategies allow widespread infection and increased viral load. Despite the efforts of immune subversion, SARS-CoV-2 infection activates innate immune pathways inducing a robust type I/III interferon response, production of proinflammatory cytokines and recruitment of neutrophils and myeloid cells. This may induce hyperinflammation or, alternatively, effectively recruit adaptive immune responses that help clear the infection and prevent reinfection. The dysregulation of the renin-angiotensin system due to down-regulation of angiotensin-converting enzyme 2, the receptor for SARS-CoV-2, together with the activation of type I/III interferon response, and inflammasome response converge to promote free radical production and oxidative stress. This exacerbates tissue damage in the respiratory system, but also leads to widespread activation of coagulation pathways leading to thrombosis. Here, we review the current knowledge of the role of the innate immune response following SARS-CoV-2 infection, much of which is based on the knowledge from SARS-CoV and other coronaviruses. Understanding how the virus subverts the initial immune response and how an aberrant innate immune response contributes to the respiratory and vascular damage in COVID-19 may help to explain factors that contribute to the variety of clinical manifestations and outcome of SARS-CoV-2 infection.
Keywords: COVID-19; SARS-CoV-2; endothelia; immunology; inflammation.
© 2020 The Authors. Clinical & Experimental Immunology published by John Wiley & Sons Ltd on behalf of British Society for Immunology.
Conflict of interest statement
D. B. and S. A. have received compensation for consultancies, presentations and advisory board activities, but no companies were involved in the decision to write and submit this manuscript. L. F. B. has nothing to disclose. S. A. has received consultancy from Novartis and Roche. D. B. has received compensation for activities related to Canbex therapeutics, InMune Biol, Lundbeck, Japan Tobacco, Merck and Novartis.
Figures



Similar articles
-
The Science Underlying COVID-19: Implications for the Cardiovascular System.Circulation. 2020 Jul 7;142(1):68-78. doi: 10.1161/CIRCULATIONAHA.120.047549. Epub 2020 Apr 15. Circulation. 2020. PMID: 32293910
-
Role of neutrophil chemoattractant CXCL5 in SARS-CoV-2 infection-induced lung inflammatory innate immune response in an in vivo hACE2 transfection mouse model.Zool Res. 2020 Nov 18;41(6):621-631. doi: 10.24272/j.issn.2095-8137.2020.118. Zool Res. 2020. PMID: 33045777 Free PMC article.
-
SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy.Theranostics. 2020 Jun 12;10(16):7448-7464. doi: 10.7150/thno.48076. eCollection 2020. Theranostics. 2020. PMID: 32642005 Free PMC article. Review.
-
Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19).J Pathol. 2020 Jul;251(3):228-248. doi: 10.1002/path.5471. Epub 2020 Jun 10. J Pathol. 2020. PMID: 32418199 Free PMC article. Review.
-
Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures.J Virol. 2020 Sep 15;94(19):e00985-20. doi: 10.1128/JVI.00985-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32699094 Free PMC article.
Cited by
-
Natural and socio-environmental factors in the transmission of COVID-19: a comprehensive analysis of epidemiology and mechanisms.BMC Public Health. 2024 Aug 13;24(1):2196. doi: 10.1186/s12889-024-19749-3. BMC Public Health. 2024. PMID: 39138466 Free PMC article. Review.
-
Advances and Challenges in COVID-19 and Pneumonia.Viruses. 2024 Feb 22;16(3):331. doi: 10.3390/v16030331. Viruses. 2024. PMID: 38543697 Free PMC article.
-
Differential Transcriptomic Landscapes of SARS-CoV-2 Variants in Multiple Organs from Infected Rhesus Macaques.Genomics Proteomics Bioinformatics. 2023 Oct;21(5):1014-1029. doi: 10.1016/j.gpb.2023.06.002. Epub 2023 Jul 13. Genomics Proteomics Bioinformatics. 2023. PMID: 37451436 Free PMC article.
-
Plausibility of natural immunomodulators in the treatment of COVID-19-A comprehensive analysis and future recommendations.Heliyon. 2023 Jun;9(6):e17478. doi: 10.1016/j.heliyon.2023.e17478. Epub 2023 Jun 21. Heliyon. 2023. PMID: 37366526 Free PMC article. Review.
-
Current hotspot and study trend of innate immunity in COVID-19: a bibliometric analysis from 2020 to 2022.Front Immunol. 2023 May 10;14:1135334. doi: 10.3389/fimmu.2023.1135334. eCollection 2023. Front Immunol. 2023. PMID: 37234160 Free PMC article.
References
-
- Collins AR. HLA class I antigen serves as a receptor for human coronavirus OC43. Immunol Invest 1993; 22:95–103. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
