

Uncovering protein structure
- PMID: 32975287
- PMCID: PMC7545034
- DOI: 10.1042/EBC20190042
Uncovering protein structure
Erratum in
-
Correction: Uncovering protein structure.Essays Biochem. 2021 Jul 26;65(2):407. doi: 10.1042/EBC-2019-0042C_COR. Essays Biochem. 2021. PMID: 34269795 Free PMC article. No abstract available.
Abstract
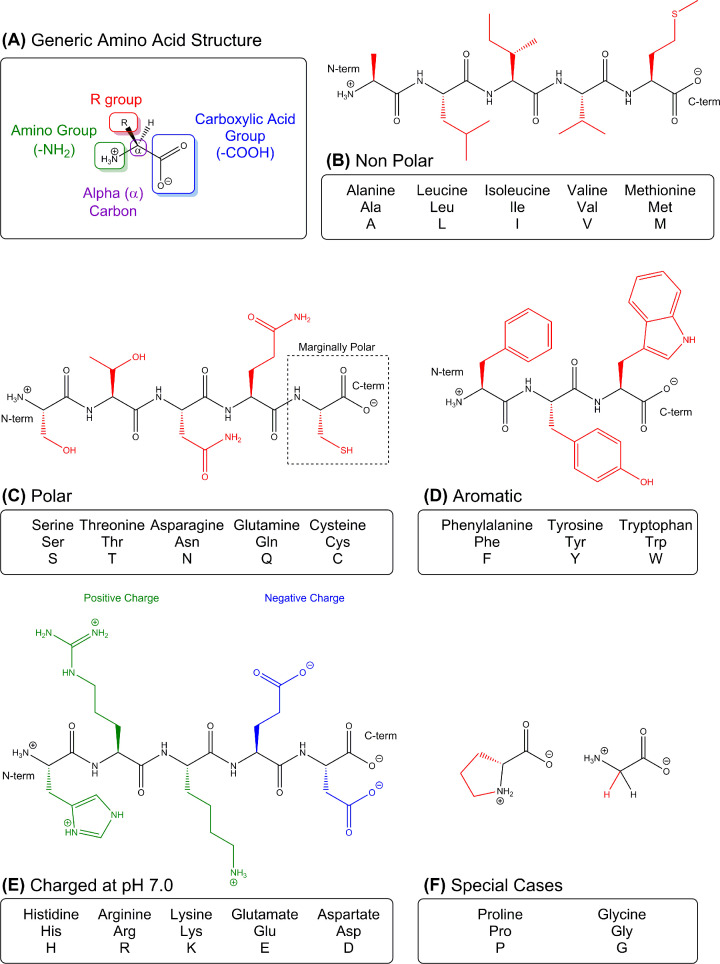
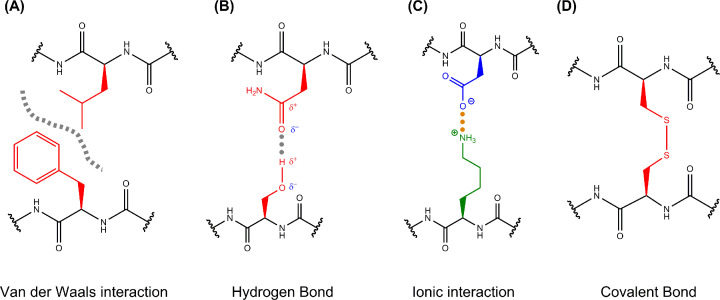
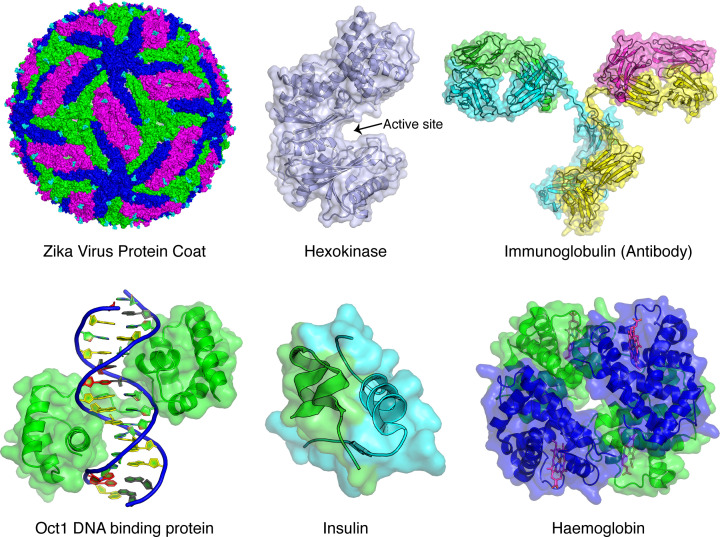
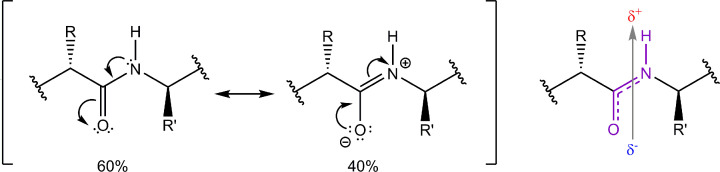
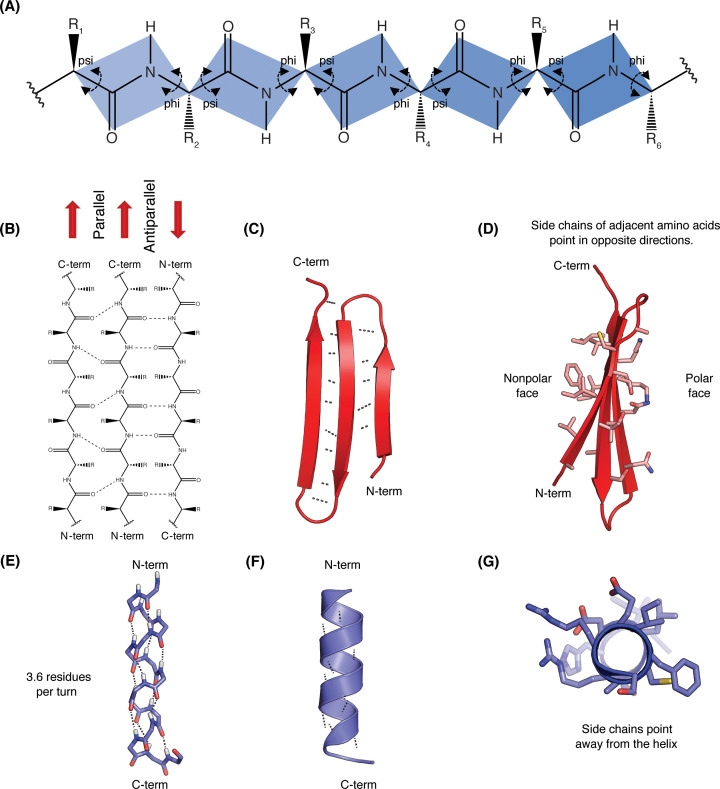
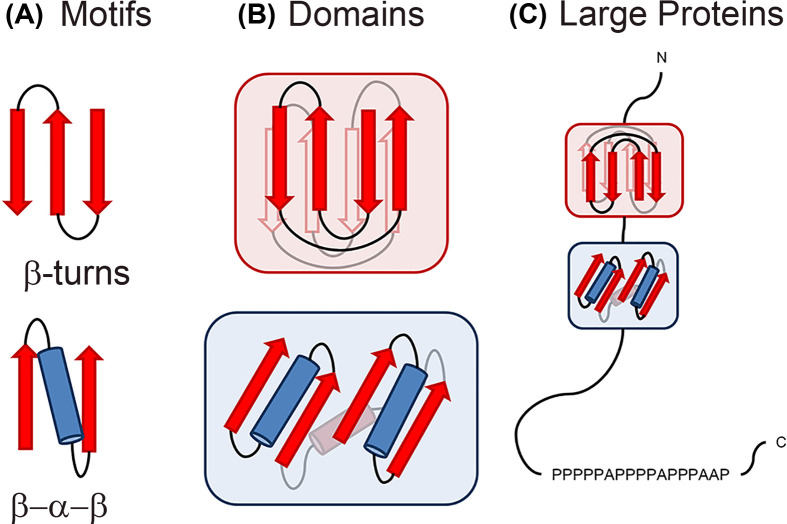
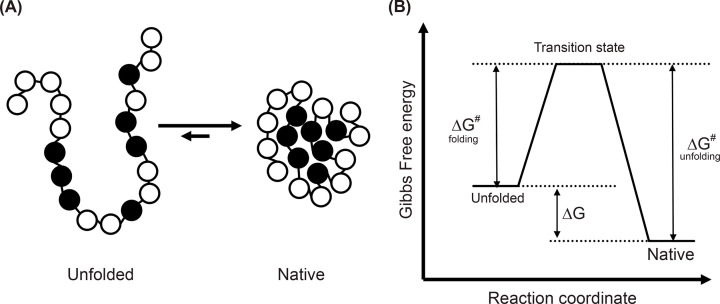
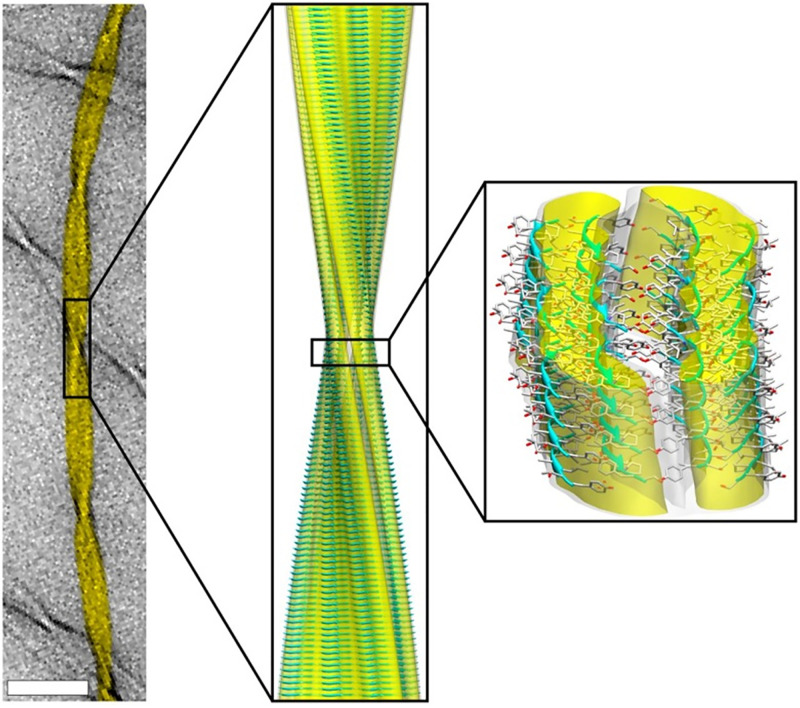
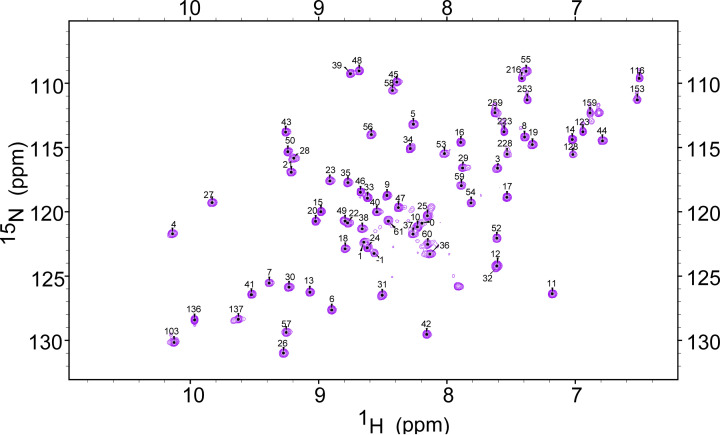
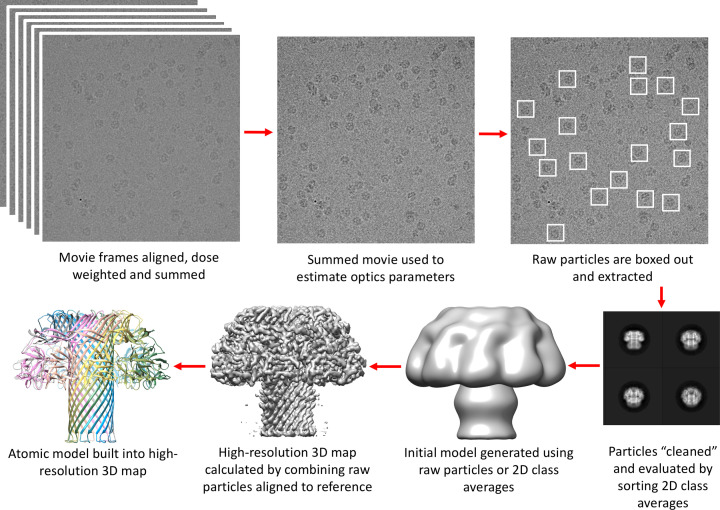
Structural biology is the study of the molecular arrangement and dynamics of biological macromolecules, particularly proteins. The resulting structures are then used to help explain how proteins function. This article gives the reader an insight into protein structure and the underlying chemistry and physics that is used to uncover protein structure. We start with the chemistry of amino acids and how they interact within, and between proteins, we also explore the four levels of protein structure and how proteins fold into discrete domains. We consider the thermodynamics of protein folding and why proteins misfold. We look at protein dynamics and how proteins can take on a range of conformations and states. In the second part of this review, we describe the variety of methods biochemists use to uncover the structure and properties of proteins that were described in the first part. Protein structural biology is a relatively new and exciting field that promises to provide atomic-level detail to more and more of the molecules that are fundamental to life processes.
Keywords: protein binding; protein chemistry; protein conformation; protein structure.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
Similar articles
-
My 65 years in protein chemistry.Q Rev Biophys. 2015 May;48(2):117-77. doi: 10.1017/S0033583514000134. Epub 2015 Apr 8. Q Rev Biophys. 2015. PMID: 25850343 Free PMC article. Review.
-
Physicochemical bases for protein folding, dynamics, and protein-ligand binding.Sci China Life Sci. 2014 Mar;57(3):287-302. doi: 10.1007/s11427-014-4617-2. Epub 2014 Feb 19. Sci China Life Sci. 2014. PMID: 24554472 Review.
-
Identifying importance of amino acids for protein folding from crystal structures.Methods Enzymol. 2003;374:616-38. doi: 10.1016/S0076-6879(03)74025-7. Methods Enzymol. 2003. PMID: 14696390 No abstract available.
-
Experimental Characterization of Protein Complex Structure, Dynamics, and Assembly.Methods Mol Biol. 2018;1764:3-27. doi: 10.1007/978-1-4939-7759-8_1. Methods Mol Biol. 2018. PMID: 29605905 Review.
-
Non-native local interactions in protein folding and stability: introducing a helical tendency in the all beta-sheet alpha-spectrin SH3 domain.J Mol Biol. 1997 May 16;268(4):760-78. doi: 10.1006/jmbi.1997.0984. J Mol Biol. 1997. PMID: 9175859
Cited by
-
Effects of non-thermal atmospheric plasma on protein.J Clin Biochem Nutr. 2022 Nov;71(3):173-184. doi: 10.3164/jcbn.22-17. Epub 2022 Sep 28. J Clin Biochem Nutr. 2022. PMID: 36447493 Free PMC article.
-
The Role of Protein Degradation in Estimation Postmortem Interval and Confirmation of Cause of Death in Forensic Pathology: A Literature Review.Int J Mol Sci. 2024 Jan 29;25(3):1659. doi: 10.3390/ijms25031659. Int J Mol Sci. 2024. PMID: 38338938 Free PMC article. Review.
-
Recent Advances in Computational Prediction of Secondary and Supersecondary Structures from Protein Sequences.Methods Mol Biol. 2025;2870:1-19. doi: 10.1007/978-1-0716-4213-9_1. Methods Mol Biol. 2025. PMID: 39543027 Review.
-
Plasma metabolomics reveals the intervention mechanism of different types of exercise on chronic unpredictable mild stress-induced depression rat model.Metab Brain Dis. 2024 Jan;39(1):1-13. doi: 10.1007/s11011-023-01310-7. Epub 2023 Nov 24. Metab Brain Dis. 2024. PMID: 37999885
-
Genomic Characteristics and E Protein Bioinformatics Analysis of JEV Isolates from South China from 2011 to 2018.Vaccines (Basel). 2022 Aug 12;10(8):1303. doi: 10.3390/vaccines10081303. Vaccines (Basel). 2022. PMID: 36016192 Free PMC article.
References
-
- Doucleff M., Hatcher-Skeers M. and Crane N.J. (2011) Pocket Guide to Biomolecular NMR, Springer
-
- Zoran P., David B., Firas K., Seth C., Jens M., Scott H. (2008) Fold your own protein, https://fold.it/
-
- David S.G., Alexander R., Maria V., Rob L. (2020) Molecular machinery: a tour of the Protein Data Bank. https://cdn.rcsb.org/pdb101/molecular-machinery/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources